RAPAMUNE*
PFIZER
Denominación genérica: Sirolimus.
Forma farmacéutica y formulación: Solución oral: sirolimus 1 mg. Excipiente csp 1 ml. Gragea: sirolimus 1mg o 2 mg. Excipiente cbp 1 gragea.
Descripción: RAPAMUNE* (sirolimus) es un agente inmunosupresor. Sirolimus es una lactona macrocíclica producida por Streptomyces hygroscopicus. El nombre químico de sirolimus es: (3S,6R,7E,9R,10R,12R,14S,15E,17E,19E, 21S,23S,26R,27R,34aS)-9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-hexadecahidro-9,27-dihidroxi -3-[(1R)-2-[(1S,3R,4R) -4-hidroxi-3-metoxiciclohexil]-1-metiletil]-10,21-dimetoxi-6,8,12,14,20,26-hexametil-23,27-epoxi-3H-pirido[2,1-c][1,4] oxaazaciclohentriacontina -1,5,11,28,29 (4H,6H,31H)-pentona. Su fórmula molecular es C51H79NO13 y su peso molecular es 914,2. La fórmula estructural de sirolimus se muestra abajo.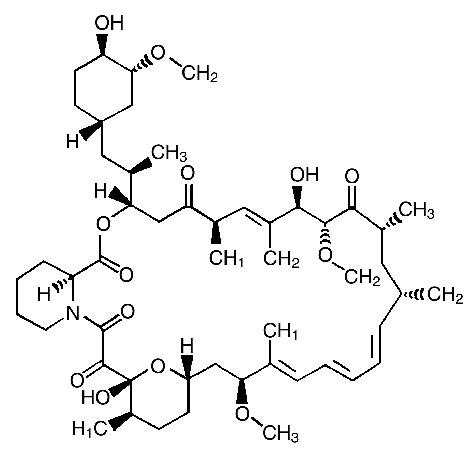
Sirolimus es un polvo blanco a blancuzco y es insoluble en agua pero muy soluble en alcohol bencílico, cloroformo, acetona, y acetonitrilo. RAPAMUNE* está disponible para su administración como una solución oral que contiene 1 mg /ml de sirolimus y también como una gragea blanca de forma triangular que contiene 1 mg o 2 mg de sirolimus.
Indicaciones terapéuticas: RAPAMUNE* está indicado para la profilaxis del rechazo de órgano en pacientes que reciben un trasplante renal. En pacientes en riesgo inmunológico de bajo a moderado, se recomienda que RAPAMUNE* sea utilizado inicialmente en un régimen con CsA y corticosteroides. CsA deberá ser retirada 2 a 4 meses después del trasplante, y la dosis de RAPAMUNE* deberá incrementarse para alcanzar la concentraciones sanguíneas recomendadas (ver Dosis y via de administración). No se ha estudiado la suspensión de ciclosporina en pacientes con rechazo agudo grado III de Banff 93 o rechazo vascular antes de suspender CsA, en pacientes dependientes de diálisis o con creatinina sérica > 4,5 mg/dl, pacientes de raza negra, retrasplantes renales, trasplantes de múltiples órganos o pacientes con un panel de títulos altos de anticuerpos relativos (ver Farmacocinética y farmacodinamia). En pacientes en alto riesgo inmunológico (definido como receptores de transplante de raza negra y/o receptores con repetición de transplante renal que perdieron un aloinjerto previo por motivos inmunológicos y/o pacientes con panel de títulos altos de anticuerpos reactivos (PRA: nivel máximo de PRA > 80%), se recomienda usar RAPAMUNE* en una combinación de tacrolimus y corticosteroides o ciclosporina y corticosteroides durante el primer año después del trasplante (ver Dosis y via de administracion y Farmacocinética y farmacodinamia). No se ha estudiado la seguridad y eficacia de estas combinaciones en pacientes de trasplante renal de alto riesgo más allá de un año. Por consiguiente, un año después del trasplante, se debe considerar cualquier ajuste al esquema inmunosupresor con base en el estado clínico del paciente.
Farmacocinética y farmacodinamia: Farmacología clínica: mecanismo de acción: sirolimus inhibe la activación y proliferación de linfocitos T que ocurre en respuesta a la estimulación por antígenos y citoquinas (interleucina [IL]-2, IL-4 e IL-15), por un mecanismo que es diferente al de otros inmunosupresores. Sirolimus también inhibe la producción de anticuerpos. En las células, sirolimus se une a la inmunofilina, proteína captadora FK-12 (FK Binding Protein-12, FKBP-12), para generar un complejo inmunosupresor. El complejo sirolimus:FKBP-12 no tiene ningún efecto sobre la actividad de la calcineurina. Este complejo se une a e inhibe la activación del blanco de rapamicina en mamíferos (mTOR; mamallian target of rapamycin), una quinasa regulatoria clave. Esta inhibición suprime la proliferación de células T dirigida por citoquinas, inhibiendo la progresión de la fase G1 a la fase S del ciclo celular. Los estudios en modelos experimentales mostraron que el sirolimus prolonga la sobrevida del aloinjerto (riñón, corazón, piel, islote, intestino delgado, pancreático-duodenal, o médula ósea) en ratones, ratas, perros, y /o primates. El sirolimus revierte el rechazo agudo del aloinjerto de corazón y riñón en ratas y prolonga la sobrevida del injerto en ratas presensibilizadas. En algunos estudios, el efecto inmunosupresor del sirolimus perdura hasta 6 meses después de la interrupción de la terapia. Este efecto de tolerancia es específico para el aloantígeno. En modelos de enfermedad autoinmune en roedores, el sirolimus suprime los eventos asociados con mediación inmune con lupus eritematoso sistémico, artritis inducida por colágeno, diabetes autoinmune tipo I, miocarditis autoinmune, encefalomielitis experimental alérgica, enfermedad injerto-versus-huésped, y uveorretinitis autoinmune. La seguridad y eficacia de RAPAMUNE* para la prevención del rechazo de órganos después del trasplante renal fue evaluada en dos estudios clínicos controlados, aleatorizados, doble-ciego, multicéntricos. Estos estudios compararon dos niveles de dosis de RAPAMUNE* (2 mg y 5 mg, una vez al día) con azatioprina o placebo cuando se administraron en combinación con CsA y corticosteroides. El estudio de RAPAMUNE* (2 mg y 5 mg, una vez al día) comparado con azatioprina fue conducido en los Estados Unidos en 38 sitios. Se enrolaron setecientos diecinueve (719) pacientes en este estudio y la aleatorización se llevó a cabo después del trasplante; 284 fueron aleatorizados para recibir 2 mg/día de RAPAMUNE*, 274 fueron aleatorizados para recibir 5 mg/día de RAPAMUNE*, y 161 para recibir azatioprina 2-3 mg/kg/día. El estudio de RAPAMUNE* (2 mg y 5 mg, una vez al día) comparado con control con placebo fue conducido en Australia, Canadá, Europa, y los Estados Unidos, con un total de 34 sitios. Se enrolaron quinientos setenta y seis (576) pacientes en este estudio y la aleatorización se realizó antes del trasplante; 227 fueron aleatorizados para recibir 2mg/día de RAPAMUNE*, 219 fueron aleatorizados para recibir 5 mg/día de RAPAMUNE*, y 130 para recibir placebo. La falla de eficacia se definió como la primera ocurrencia de un episodio agudo de rechazo (confirmado por biopsia), pérdida del injerto, o muerte. Los análisis primarios de eficacia de estos estudios determinaron que RAPAMUNE*, a dosis de 2mg/día y 5 mg/día, redujeron significativamente la incidencia del fallo de eficacia a los seis meses después del trasplante comparado con ambos azatioprina y placebo. La reducción en la incidencia del primer episodio de rechazo agudo confirmado con biopsia en los pacientes tratados con RAPAMUNE* comparado con los grupos de control incluyó una reducción en todos los grados de rechazo. Las tasas se sobrevivencia del injerto y del paciente, que eran los objetivos co-primarios, fueron similares en los pacientes tratados con RAPAMUNE* y tratados con comparador a un año. La tabla abajo resume los resultados de los análisis primarios de eficacia de estos estudios. RAPAMUNE* Solución oral, a dosis de 2 mg/día y 5 mg/día, reduce significativamente la incidencia de un fallo de eficacia (estadísticamente significativo a un nivel de < 0,025; el nivel nominal de significancia ajustado para comparaciones múltiples de dosis [2]) a seis meses después del trasplante comparado con ambos azatioprina y placebo.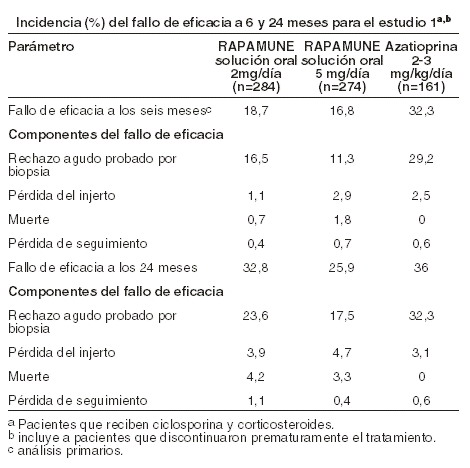
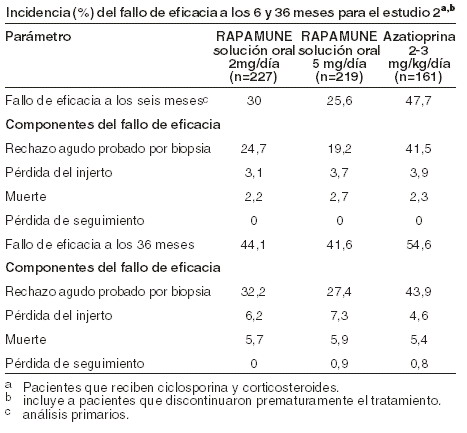
La sobrevida del injerto y del paciente a un año fue el coanálisis primario. La siguiente tabla muestra la sobrevida del injerto y pacientes a uno y dos años en el Estudio 1 y 1 y 3 años del Estudio 2. Las tasas de sobrevida del injerto y el paciente fueron similares en pacientes tratados con RAPAMUNE* y con el comparador.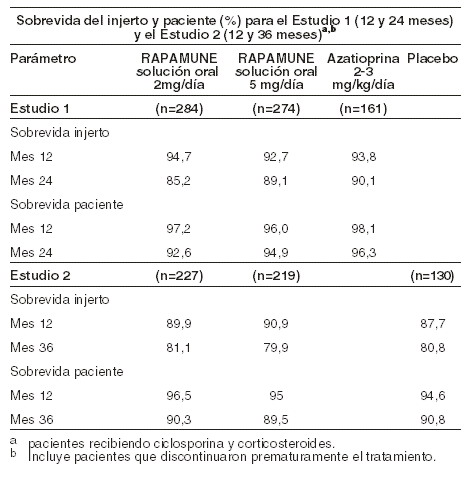
La reducción en la incidencia de los episodios de rechazo agudo primario confirmados por biopsia en pacientes tratados con RAPAMUNE* comparados con los grupos control incluida una reducción en todos los grados de rechazo. El estudio de RAPAMUNE* (2 mg y 5mg, una vez al día) comparado con azatioprina que fue estratificado prospectivamente por raza dentro del centro de estudio, el fallo de eficacia fue similar para el RAPAMUNE* 2 mg/día y menor para el RAPAMUNE* 5 mg/día comparado con azatioprina en pacientes negros. El estudio de RAPAMUNE* controlado con placebo (2 mg y 5 mg, una vez al día) el cual no fue estratificado prospectivamente por raza, el fallo de eficacia fue similar para ambas dosis de RAPAMUNE* comparadas al placebo en pacientes de raza negra.
Las tasas de filtración glomerular media (TFG) a un año post-trasplante fueron calculadas utilizando la ecuación de Nankivell para todos los sujetos en ambos estudios que tuvieron creatinina sérica medida a los 12 meses. En los Estudios 1 y 2, la media TFG a un año fue menor en pacientes tratados con CsA y RAPAMUNE* comparados con aquellos tratados con CsA y el control respectivo con azatioprina o placebo. Dentro de cada grupo de tratamiento para ambos estudios, la media TFG a un año post-trasplante fue menor en pacientes que experimentaron por lo menos un episodio de rechazo agudo comprobado por biopsia, comparado con aquellos que no lo presentaron. Se evaluaron la seguridad y eficacia de RAPAMUNE* como esquema de mantenimiento después de suspender CsA, 3 a 4 meses después del transplante renal. Se reclutaron quinientos veinticinco (525) pacientes en un estudio aleatorizado, multicéntrico, controlado, realizado en 57 centros en Australia, Canadá y Europa. Todos los pacientes de este estudio recibieron la formulación en gragea. Este estudio comparó pacientes a quienes se les administró RAPAMUNE*, CsA y corticosteroides en forma continua, con pacientes que recibieron el mismo tratamiento convencional durante los primeros 3 meses después del trasplante (período previo a la aleatorización) y después se suspendió CsA. Durante el retiro de CsA, las dosis de RAPAMUNE* se ajustaron para lograr los rangos objetivo de concentración mínima en sangre total de sirolimus (16 a 24 ng/ml hasta el mes 12, después 12 a 20 ng/ml desde ahí hasta el mes 60). A los 3 meses, 430 pacientes fueron distribuidos en forma aleatoria en partes iguales, ya sea al tratamiento de RAPAMUNE* con CsA, o RAPAMUNE* como esquema de mantenimiento después de suspender CsA. La elegibilidad para la aleatorización incluyó: ningún episodio de rechazo agudo Banff Grado 3 o rechazo vascular en las 4 semanas anteriores a la asignación aleatoria; creatinina sérica ≤ 4,5 mg/dl y una función renal adecuada para soportar el retiro de CsA (en opinión del investigador). El objetivo final primario de eficacia fue sobrevida del injerto 12 meses después del trasplante. Los objetivos finales secundarios de eficacia fueron las tasas de rechazo agudo confirmadas por biopsia, sobrevida del paciente, incidencia de falla de la eficacia (definida como lo primero que se presente, ya sea rechazo agudo comprobado por biopsia, pérdida del injerto o muerte), y falla de tratamiento (definido como lo primero que se presente, ya sea abandono, rechazo agudo, pérdida del injerto o muerte). La siguiente tabla resume la sobrevida del injerto y del paciente a los 12, 24, 36 y 48 meses de este estudio. A los 12, 24 y 36 meses, la sobrevida del injerto y del paciente fueron similares para ambos grupos. A los 48 meses, la sobrevida del paciente también fue similar para ambos grupos, pero la diferencia de la sobrevida del injerto entre los dos grupos alcanzó una significancia estadística.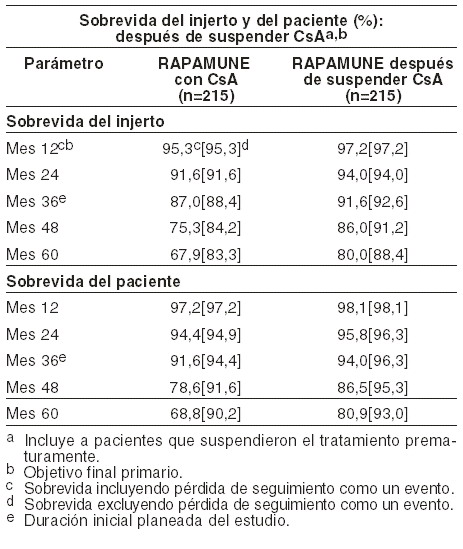
La siguiente tabla resume los resultados del primer rechazo agudo comprobado por biopsia a los 12 y 60 meses. Hubo una diferencia significativa en el primer rechazo comprobado por biopsia entre los dos grupos durante el período posterior a la aleatorización hasta los 12 meses. Sin embargo, al mes 60, la diferencia entre los dos grupos no fue significativa (6,5% vs. 10,2%, respectivamente). La mayoría de los rechazos agudos posteriores a la aleatorización se presentaron en los primeros 3 meses después de la aleatorización.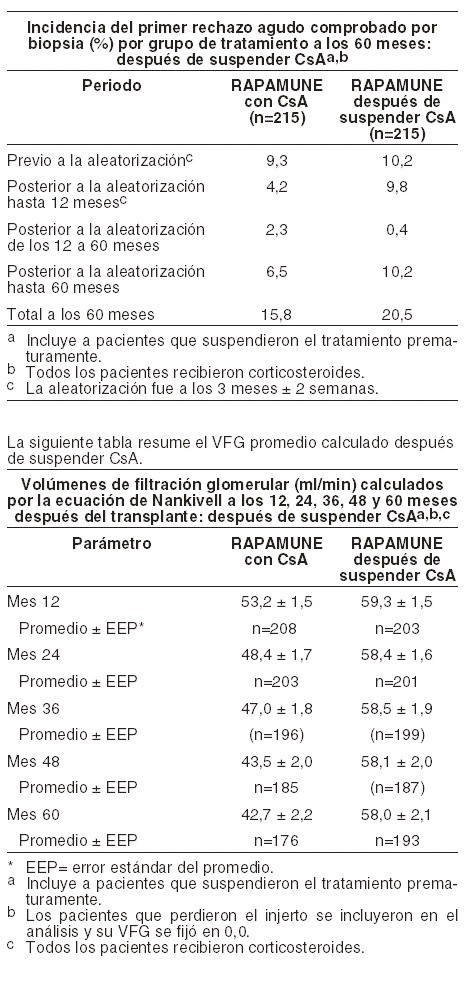
El VFG promedio a los 12, 24, 36, 48 y 60 meses calculado por la ecuación de Nankivell, fue significativamente más alto para los pacientes que recibieron RAPAMUNE* como esquema de mantenimiento después de suspender CsA que para los pacientes en el grupo de tratamiento de RAPAMUNE* con CsA. Al mes 60, los pacientes con rechazo agudo en cualquier momento después del trasplante, tuvieron una VFG promedio calculada significativamente más alta en pacientes que recibieron RAPAMUNE* como tratamiento de mantenimiento después de suspender CsA que los pacientes en el grupo de tratamiento RAPAMUNE* con CsA. La seguridad y eficacia de la conversión de los inhibidores de calcineurina (ICN) a RAPAMUNE* fueron valoradas en los pacientes con trasplante renal en mantenimiento. Este estudio fue aleatorizado, multicéntrico, estudio controlado conducido en 111 centros globales, incluyendo Estados Unidos y Europa. Se enrolaron ochocientos treinta pacientes (830) estratificados por tasa calculada basal de filtración glomerular (TFG, 20-40 ml/min vs. mayor de 40 ml/min. El enrolamiento en el estrato de pacientes con TFG basal calculado menor a 40 ml/min fue discontinuado debido a una falta de balance en los eventos de seguridad (ver Precauciones generales y Reacciones secundarias y adversas). Este estudio comparó pacientes trasplantados renales (6-120 meses después del trasplante) que fueron convertidos de inhibidores de calcineurina a RAPAMUNE*, con pacientes que continuaron recibiendo inhibidores de calcineurina. Se inició medicación inmunosupresora concomitante, incluyendo micofenolato de mofetilo (MMF), azatioprina (AZA), y corticosteroides. RAPAMUNE* fue iniciado con una sola dosis de carga de 12-20 mg, después de la cual la dosis fue ajustada para alcanzar un objetivo de concentración mínima de sirolimus en sangre completa de 8-20 ng/ml (método cromatográfico). El punto final primario de eficacia fue la TFG calculada a los 12 meses post-aleatorización. Los puntos finales secundarios incluyeron rechazo agudo confirmado por biopsia, pérdida del injerto, y muerte. Se resumen abajo los hallazgos en el estrato de pacientes con TFG basal calculada mayor de 40 ml/min (conversión de RAPAMUNE*, n=497; continuación ICN, n=246): no hubo una mejoría estadísticamente o clínicamente significativa en el TFG Nankivell comparado con el análisis primario.
En el estrato de pacientes con una TFG basal calculada mayor de 40 ml/min (conversión de RAPAMUNE*, n=497; continuación de ICN, n=246), la función renal y las tasas de rechazo agudo, pérdida del injerto, y muerte fueron muy similares a 1 y 2 años. Ocurrieron más frecuentemente los eventos adversos tratamiento-emergentes durante los primeros seis meses después de la conversión de RAPAMUNE*. Las tasas de neumonía fueron significativamente mayores en el grupo de conversión de sirolimus. Mientras que los valores de la media y la mediana de la tasa de proteína urinaria a creatinina fueron similares entre los grupos de tratamiento en el análisis primario, se observaron valores significativamente mayores de la media y mediana de la excreción de la proteína urinaria en el brazo de conversión de RAPAMUNE* a un año y a dos años, como se muestra en la tabla abajo. Además, cuando se comparó a los pacientes que continuaron recibiendo inhibidores de calcineurina, un mayor porcentaje de pacientes tuvieron una tasa de proteína urinaria a creatinina > 1 a uno - dos años después de la conversión de sirolimus. Esta diferencia se vio en ambos pacientes los que tuvieron una tasa de proteína urinaria a creatinina > 1 y aquellos que tuvieron una tasa de proteína a creatinina > 1 en el análisis primario. Más pacientes en el grupo de conversión de sirolimus desarrollaron proteinuria de rango nefrótico, como se define por una tasa de proteína urinaria a creatinina > 3,5 (46/482 [9,5%] vs. 9/239 [3,8%]), aun cuando los pacientes con proteinuria de rango nefrótico de análisis primario fueron excluidos. La tasa de proteinuria de rango nefrótico fue significativamente mayor en el grupo de conversión de sirolimus comparado con el grupo de continuación con inhibidor de calcineurina con un análisis primario de tasa de proteína urinaria a creatinina > 1 (13/29 vs 1/14), excluyendo los pacientes con análisis primario de proteinuria de rango nefrótico.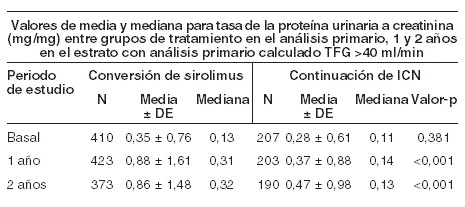
La información de arriba deberá de tomarse en consideración cuando se considere la conversión de los inhibidores de calcineurina a RAPAMUNE* en pacientes trasplantados renales estables debido a pérdida de evidencia, mostrando que la función renal mejora después de la conversión, y el hallazgo de un mayor incremento en la excreción de proteína urinaria, y un incremento en la incidencia de proteinuria de tratamiento-emergente de rango nefrótico después de la conversión a RAPAMUNE*. Esto fue particularmente verdadero entre los pacientes con excreción de proteína urinaria anormal existente antes de la conversión. En el estrato con una TFG basal calculada mayor de 40 ml/min, los valores de la media y mediana de la tasa de proteína urinaria a creatinina fueron similares entre los grupos de tratamiento al nivel basal (media 0,35 y 0,28; mediana 0,13 y 0,11 para la conversión de RAPAMUNE* y grupos de continuación de ICN, respectivamente). A los 24 meses, la media y mediana de la tasa de proteína urinaria a de creatinina fueron significativamente mayores en el grupo de conversión de RAPAMUNE* como se comparó con aquellos grupos de (ICN) (media: 0,87 y 0,48, p < 0,0002; mediana 0,33 y 0,13, p < 0,001, para los grupos de conversión de RAPAMUNE* y grupos de continuación de ICN, respectivamente)[ver Precauciones generales]. Se reportó también (síndrome nefrótico) de inicio (ver Reacciones secundarias). A los 2 años, la tasa de neoplasias de piel no-melanoma fue significativamente menor en el grupo de conversión de RAPAMUNE* comparado con el grupo de continuación de ICN (1,8% y 6,9%, respectivamente, p < 0,001). Esta diferencia en las tasas de las neoplasias en piel persistió después de la exclusión de pacientes con una historia previa de neoplasias de piel (0,7% y 4,1% para los grupos de conversión de RAPAMUNE* y ICN, respectivamente, p < 0,002). Se deberá de notar que el estudio 4 no fue diseñado para considerar los factores de riesgo de neoplasias o identificación sistemática de sujetos en busca de neoplasias. En un subgrupo de pacientes en estudio con una TFG basal mayor de 40 ml/min y excreción de proteína urinaria normal, la TFG calculada fue mayor al año y dos años en pacientes convertidos a RAPAMUNE* (n=197) que para el subgrupo correspondiente de pacientes de continuación de ICN (n=102). Las tasas de rechazo agudo, pérdida de injerto, y muerte fueron similares, pero la excreción de proteína urinaria fue incrementada en el brazo de tratamiento de RAPAMUNE* del subgrupo. RAPAMUNE* se estudió en un estudio clínico aleatorizado, abierto, controlado de un año en pacientes de alto riesgo definidos como receptores de transplante de raza negra y/o receptores con repetición de trasplante renal que perdieron un aloinjerto previo por motivos inmunológicos y/o pacientes con panel de títulos altos de anticuerpos reactivos (PRA: nivel máximo de PRA > 80%). Los pacientes se aleatorizaron en una proporción de 1:1 para concentración controlada de sirolimus y tacrolimus o concentración controlada de sirolimus y ciclosporina (modificada), y ambos grupos recibieron corticosteroides según la práctica local. El protocolo permitió la inducción de anticuerpos como se definió prospectivamente en cada centro de trasplante, y se usó en 85,3% de los pacientes. El estudio se realizó en 35 centros de Estados Unidos. La demografía al inicio estaba bien equilibrada en ambos grupos; el porcentaje de pacientes de raza negra era del 77,7% en el grupo de sirolimus y tacrolimus y del 77,2% en el de sirolimus y ciclosporina. La población evaluable por intención de tratar (definida como todos los pacientes que fueron aleatorizados y que recibieron un trasplante y por lo menos una dosis del medicamento del estudio) incluyó 224 pacientes que recibieron sirolimus y tacrolimus y 224 pacientes que recibieron sirolimus y ciclosporina. Los puntos finales coprimarios medidos a los 12 meses en la población IdT, fueron fracaso de eficacia (definida como la primera aparición de rechazo agudo confirmada por biopsia, pérdida del injerto o muerte), primera pérdida del injerto o muerte y función renal medida por el IFG calculado por la fórmula de Nankivell. La siguiente tabla resume los puntos finales coprimarios. Las tasas globales de fracaso de eficacia y la primera pérdida del injerto o muerte fueron similares en ambos grupos.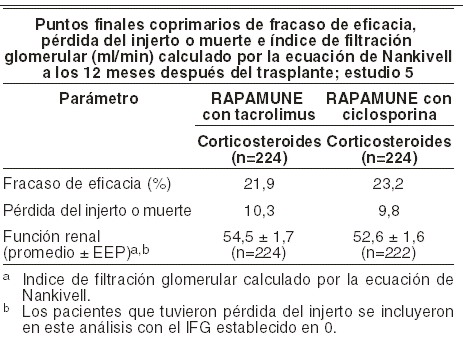
La sobrevida a 12 meses fue de 95,1% en pacientes que recibieron sirolimus y tacrolimus vs 94,6% en pacientes que recibieron sirolimus y ciclosporina. La incidencia de rechazo agudo confirmado por biopsia fue de 13,8% en pacientes que recibieron sirolimus y tacrolimus vs 17,4% en pacientes que recibieron sirolimus y ciclosporina. A pesar de que el rechazo agudo fue numéricamente más bajo en pacientes que recibieron sirolimus y tacrolimus, la severidad del rechazo fue estadísticamente mayor comparada con la de los pacientes que recibieron sirolimus y ciclosporina. La función renal bajo tratamiento fue consistentemente más alta en pacientes que recibieron sirolimus y tacrolimus comparada con pacientes que recibieron sirolimus y ciclosporina. Un estudio clínico en pacientes con trasplante hepático aleatorizados de un régimen basado-INC al régimen sirolimus-basado contra la continuación de un régimen de INC-basado de 6-144 meses post-trasplante hepático falló para demostrar la superioridad en el RFG ajustado basal a los 12 meses (-4,45 ml/min y -3,07 ml/min, respectivamente). El estudio también falló para demostrar la tasa de no inferioridad de la pérdida del injerto combinada, datos de sobrevida faltantes, o muerte para el grupo de conversión de sirolimus comparado con el grupo de continuación INC. El número de muertes en el grupo de conversión de sirolimus fue mayor que en el grupo de continuación INC, aunque la diferencia no fue estadísticamente significativa. Las tasas de discontinuación prematura del estudio, eventos adversos globales (e infecciones, específicamente), y rechazo de injerto hepático agudo biopsia-probada a los 12 meses fueron significativamente mayores en el grupo de conversión de sirolimus comparado con el grupo de continuación de INC. RAPAMUNE* fue evaluado en un estudio clínico a 36 meses, abierto, aleatorizado, controlado en 14 centros de Norteamérica en receptores pediátricos de trasplante renal (edad 3 a < 18 años) considerados a un riesgo inmunológico alto para desarrollar nefropatía alógrafa crónica, definida como una historia de uno o más episodios de rechazos agudos y/o la presencia de nefropatía crónica en la biopsia renal. Setenta y ocho (78) pacientes fueron aleatorizados en una proporción de 2:1 de RAPAMUNE* (concentraciones objetivo de sirolimus de 5 a 15 ng/ml, por análisis cromatográfico, n=53) en combinación con un inhibidor de calcineurina y corticosteroides o continuar con la terapia inmunosupresora basada en inhibidor de calcineurina (n=25). El punto final primario del estudio fue la falla en la eficacia definida como la primera aparición de una biopsia confirmada de rechazo agudo, pérdida del injerto, o muerte, y el estudio se designó para demostrar la superioridad de RAPAMUNE* añadido a un régimen inmunosupresor basado en inhibidor de calcineurina comparado con un régimen basado en inhibidor de calcineurina. El índice acumulativo hasta 36 meses del fallo en la eficacia fue de 45,3% en el grupo de RAPAMUNE* comparado con 44% en el grupo de control, y no demostró superioridad. Hubo una muerte en cada grupo. El uso de RAPAMUNE* en combinación con inhibidores de calcineurina y corticosteroides se asoció con un incremento en el riesgo de deterioro de la función renal, anomalías en los lípidos séricos (incluyendo pero no limitado a un incremento en los triglicéridos séricos y colesterol), e infecciones del tracto urinario. Este estudio no da apoyo a la adición de RAPAMUNE* en la terapia inmunosupresora basada en inhibidor de calcineurina en esta subpoblación de pacientes pediátricos con trasplante renal. (ver Dosis y via de administracion). Farmacocinética: absorción: luego de la administración de la solución oral, RAPAMUNE* se absorbe rápidamente, con un tiempo medio a la concentración pico (tmáx) de aproximadamente 1 hora después de una dosis única en sujetos sanos, y de aproximadamente 2 horas después de dosis múltiples en receptores de trasplante renal. Después de la administración de grageas, la tmáx de RAPAMUNE* fue de aproximadamente 3 horas después de dosis unitarias en voluntarios sanos y múltiples dosis en pacientes con transplante renal. Se estimó que la disponibilidad sistémica (F) de RAPAMUNE* de la solución oral es de aproximadamente 14%. Después de la administración de grageas, se estimó que F era aproximadamente 17%. La bioequivalencia entre grageas de 1 mg y 2 mg se ha mostrado generalmente en voluntarios sanos. Las concentraciones de RAPAMUNE* son proporcionales a la dosis entre 3 y 12 mg /m2 después de la administración de solución oral en pacientes con trasplante renal estable, y entre 5 y 40 mg después de la administración de grageas en voluntarios sanos. Efectos de los alimentos: en 22 voluntarios sanos, que recibieron sirolimus en solución oral, un desayuno (860 kcal, 55% kcal de grasa) alto en grasa alteró las características de biodisponibilidad de RAPAMUNE*. Comparando con condiciones de ayuno, se observó una disminución de 34% en la concentración sanguínea máxima de sirolimus (Cmáx), un aumento de 3,5 veces en el tiempo a la concentración máxima (tmáx), y un aumento de 35% en la exposición total media (ABC). En un estudio idéntico, RAPAMUNE* fue administrado en grageas a 24 sujetos sanos. Los valores para Cmáx, tmáx, y ABC mostraron incrementos de 65%, 32%, y 23%, respectivamente. Así, un alimento alto en grasas produjo diferencia en las dos formulaciones con respecto a la tasa de absorción, pero no a la extensión de la absorción. La evidencia de un gran estudio multicéntrico aleatorizado controlado comparando RAPAMUNE* Solución oral con las grageas, apoya que las diferencias en las tasas de absorción no afectan la eficacia del medicamento. Para minimizar la variabilidad en los niveles sanguíneos, RAPAMUNE* deberá ser tomado consistentemente con o sin alimentos. Las pruebas de bioequivalencia basadas en la exposición total (ABC) y la concentración sanguínea máxima (Cmáx) mostraron que RAPAMUNE* administrado con jugo de naranja es equivalente a su administración con agua. Por lo que, el jugo de naranja y el agua pueden ser utilizados de manera intercambiable para diluir RAPAMUNE* Solución oral. El jugo de toronja reduce el metabolismo del medicamento mediado por CYP3A4 y potencialmente incrementa el transporte en contra de medicamento desde los enterocitos del intestino delgado mediado por P-gp, y no debe ser usado para dilución o tomado junto con RAPAMUNE* (ver Interacciones medicamentosas y de otro género). Distribución: la proporción media (±DEs) sangre - a - plasma de RAPAMUNE* fue de 36 (±17,9) en receptores estables de aloinjerto renal, después de la administración de la solución oral, indicando que sirolimus es extensamente distribuido hacia los elementos formes de la sangre. El volumen medio de distribución (Vee /F) de RAPAMUNE* Solución oral es de 12 + 7,52 L /kg. Sirolimus se une extensamente (aproximadamente 92%) a las proteínas plasmáticas humanas. En sangre total en el hombre, se demostró que la unión de sirolimus está asociada principalmente con la albúmina sérica (97%), la glucoproteína ácida 1, y las lipoproteínas. Metabolismo: sirolimus es un sustrato tanto para el citocromo CYP3A4 como para la glucoproteína P. Sirolimus es extensamente metabolizado por O-demetilación y/o hidroxilación. Siete (7) metabolitos principales, incluyendo los derivados hidroxi, demetil, e hidroxidemetil, son identificables en sangre total. Algunos de estos metabolitos también son detectables en muestras plasmáticas, fecales y urinarias. Los conjugados glucurónido y sulfato no están presentes en ninguna de las matrices biológicas. Sirolimus es el principal componente en sangre total humana, y contribuye con más del 90% de la actividad inmunosupresora. Excreción: después de una dosis única de [14C] sirolimus a sujetos sanos, la mayor parte (91%) de la radioactividad fue recuperada de las heces, y solamente una cantidad menor (2,2%) fue excretada en la orina. La media DE de la vida media terminal de eliminación (t1/2) de RAPAMUNE* después de administración de dosis múltiples de solución oral en pacientes estables con trasplante renal fue estimada en aproximadamente 62±16 horas. Pacientes con trasplante renal: los parámetros farmacocinéticos medios (±DE) para RAPAMUNE* administrado en solución oral diariamente en combinación con CsA y corticosteroides en pacientes con trasplante renal fueron determinados en los meses 1, 3, y 6 después del trasplante. No hubo diferencias significativas en Cmáx, tmáx, ABC o Dep /F con respecto al grupo de tratamiento o al mes. Después de la administración diaria de solución oral y grageas de RAPAMUNE* en pacientes con trasplante renal, no parecieron ser diferentes los estimados de Cmáx, ABC, y Dep/F, pero la tmáx fue significativamente diferente. Luego de una administración de RAPAMUNE* Solución oral dos veces al día repetida sin dosis inicial de carga en un estudio de dosis múltiples, la concentración mínima promedio de sirolimus se aumentó aproximadamente 2 a 3 veces a lo largo de los primeros 6 días de tratamiento, en cuyo momento se alcanza el estado de equilibrio. La concentración mínima media en sangre completa de sirolimus en pacientes que reciben RAPAMUNE* en solución oral o grageas con una dosis de carga de tres veces la dosis de mantenimiento alcanzaron las concentraciones de estado estable dentro de las 24 horas después de iniciar la administración de la dosis. Pacientes de alto riesgo: la siguiente tabla resume las dosis promedio de RAPAMUNE* y las concentraciones mínimas promedio en sangre completa para las grageas administradas todos los días en combinación con ciclosporina o tacrolimus, y corticosteroides en pacientes con trasplante renal en alto riesgo (ver Farmacodinamia).
Los pacientes tratados con la combinación de RAPAMUNE* y tacrolimus requirieron dosis mayores de RAPAMUNE* para lograr las concentraciones objetivo de sirolimus que los pacientes tratados con la combinación de RAPAMUNE* y ciclosporina. Se resumen en la siguiente tabla los parámetros farmacocinéticos de sirolimus en pacientes adultos con trasplante renal después de múltiples dosis con RAPAMUNE* 2 mg diariamente, en combinación con ciclosporina y corticosteroides.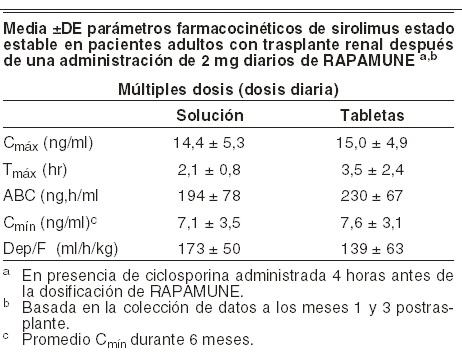
Fue correlacionada significativamente con ABCt, ss las concentraciones de sirolimus mínimas de sangre completa, medida por CL/EM/EM en pacientes trasplantados renales. Una vez repetida, la administración dos veces al día sin una carga de dosis inicial en un estudio de dosis múltiple, el promedio mínimo de la concentración de sirolimus se incrementa aproximadamente 2 a 3 veces sobre la terapia inicial de 6 días, a la cual se alcanza el estado estable. Una dosis de carga de 3 veces la dosis de mantenimiento proporcionará concentraciones cercanas al estado estable en un día en la mayoría de los pacientes.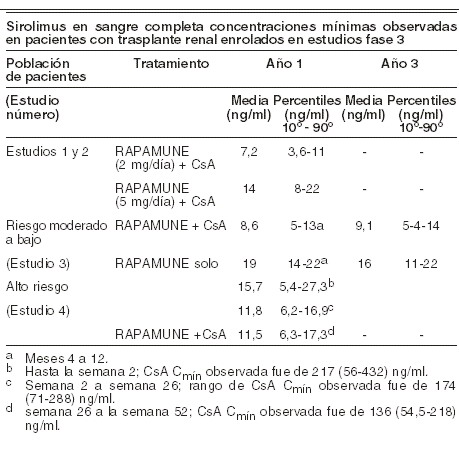
El retiro de la ciclosporina y los incrementos concurrentes en las concentraciones mínimas de sirolimus para el estado estable requirieron aproximadamente de 6 semanas. Después del retiro de la ciclosporina, se requirieron dosis mayores de RAPAMUNE* debido a la ausencia de la inhibición del metabolismo de sirolimus y el trasporte por ciclosporina y al alcanzar objetivos mayores de concentraciones mínimas de sirolimus durante la administración de concentraciones controladas. Poblaciones especiales: deterioro hepático: RAPAMUNE* (15 mg) fue administrado como una dosis oral única en solución oral a sujetos con función hepática normal y a pacientes con una clasificación Child-Pugh de deterioro hepático grado A (medio), o B (moderado) o C (severo). Comparado con los valores del grupo de función hepática normal, los pacientes con deterioro hepático medio, moderado, y severo tuvieron 43%, 94% y 189% valores medios más altos para el ABC y el t1/2de sirolimus, y tuvo valores medios más bajos para la Dep/F d CsA Cmín observada de sirolimus. La tasa de absorción de sirolimus no fue afectada por la enfermedad hepática, como fue evidenciado por los valores de Cmax y tmáx. La dosis de mantenimiento de RAPAMUNE* deberá de ser reducida en aproximadamente un tercio en pacientes con deterioro hepático medio a moderado y por aproximadamente una mitad en pacientes con daño hepático severo basado en una disminución en la depuración (ver Dosis y via de administracion). En pacientes con daño hepático, se necesita que se realice control de niveles mínimos de sirolimus en sangre completa. En pacientes con deterioro hepático severo, se debe tener en consideración el monitoreo cada 5 a 7 días por un período mayor de tiempo después del ajuste de dosis o después de la dosis de carga, debido a un retraso para alcanzar el estado estable debido a la vida media prolongada. Deterioro renal: hay una excreción renal mínima del fármaco o sus metabolitos. La farmacocinética de sirolimus es muy similar en varias poblaciones con función renal en un rango de normal a ausente (pacientes en diálisis). Pediátrica: se recolectaron los datos farmacocinéticos de sirolimus en estudios controlados-concentración de pacientes pediátricos con trasplante renal que también recibían ciclosporina y corticosteroides. Los rangos objetivo para concentraciones mínimas fueron 10-20 ng/ml para 21 niños recibiendo grageas, o 5-15 ng/ml para un niño recibiendo solución oral. Los niños de una edad de 6-11 años (n=8) recibieron media ±dosis de DEs de 1,75±0,71 mg/día (0,064±0,018 mg/kg, 1,65±0,43 mg/mg2). Los niños de una edad de 12-18 años (n=14) recibieron media dosis de DEs de 2,79±1,25 mg/día (0,053 0,0150 mg/kg, 1,86±0,61 mg/mg2). Al momento del muestreo de sangre con sirolimus para la evaluación farmacocinética, la mayoría (80%) de estos pacientes pediátricos recibieron la dosis de sirolimus a las 16 horas después de la dosis diaria única de ciclosporina.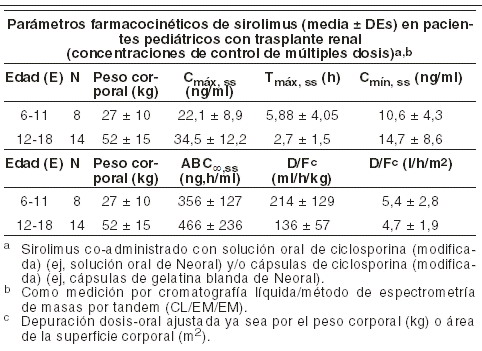
La tabla abajo resume los datos farmacocinéticos obtenidos en pacientes pediátricos bajo diálisis con función renal crónicamente deteriorada recibiendo solución oral de RAPAMUNE*.
Geriátrica: los estudios clínicos de RAPAMUNE* no incluyeron un número suficiente de pacientes > de 65 años de edad para determinar si responderán en forma diferente de los pacientes más jóvenes. Los datos sobre la concentración mínima de RAPAMUNE* después de recibir solución oral en 35 pacientes de trasplante renal > 65 años de edad fueron similares a los de la población adulta (n = 822) de 18 a 65 años de edad. Género: la depuración de dosis oral en solución oral de RAPAMUNE* en hombres fue 12% más baja que la de las mujeres; los sujetos masculinos tuvieron un t1/2significativamente más largo que las sujetos femeninos (72,3 horas versus 61,3 horas). Se obtuvieron efectos similares de género y depuración de la dosis oral y t1/2 después de la administración de RAPAMUNE* en grageas. Estas diferencias farmacocinéticas no requieren ajustes de dosis con base en el género. Raza: en estudios grandes de Fase III que emplearon RAPAMUNE* y ciclosporina microemulsión solución oral (modificado) y /o cápsulas de ciclosporina (modificado), no hubo diferencias significativas en las concentraciones mínimas medias de sirolimus a lo largo del tiempo entre pacientes de raza negra (n = 139) y no negra (n = 724), durante los primeros 6 meses después del trasplante, a dosis de RAPAMUNE* de 2 mg/día y 5 mg/día en solución oral.
Contraindicaciones: RAPAMUNE* está contraindicado en pacientes con hipersensibilidad a sirolimus o sus derivados, o a cualquier componente del producto farmacéutico.
Precauciones generales: RAPAMUNE* está destinado a la administración oral solamente. Curación de heridas y acumulación de líquidos: los inhibidores mTOR como el sirolimus han mostrado in vitro inhibir la producción de ciertos factores de crecimiento que podrían afectar la angiogénesis, proliferación de fibroblastos, y permeabilidad vascular. Han habido reportes de curación de heridas tardía o alterada en pacientes recibiendo RAPAMUNE*, incluyendo linfocele y dehiscencia de las heridas. El linfocele, una complicación quirúrgica conocida del trasplante renal, ocurrió significativamente más frecuentemente en una manera relacionada con la dosis en pacientes tratados con RAPAMUNE*. Deberán considerarse medidas adecuadas para minimizar tales complicaciones. Los pacientes con una IMC mayor de 30 kg/m2 podrían estar en un riesgo incrementado de curación de heridas alterado basados en los datos de la literatura médica (ver Reacciones secundarias y adversas). En pacientes recibiendo RAPAMUNE* han habido también reportes de acumulación de líquidos, incluyendo edema periférico, linfoedema, efusión pleural y efusiones pericárdicas (incluyendo en niños y adultos efusiones hemodinámicamente significativas). Malignidad en piel: la inmunosupresión incrementa la susceptibilidad para desarrollar linfoma y otras enfermedades malignas, particularmente de la piel. Por lo tanto, los pacientes que estén tomando RAPAMUNE* deben limitar su exposición a la luz solar y luz ultravioleta utilizando ropa protectora y filtro solar con un alto factor de protección (Ver Reacciones secundarias y adversas). Hiperlipidemia: el uso de RAPAMUNE* puede llevar a incrementar el colesterol sérico y los triglicéridos que puede requerir tratamiento. Los pacientes deben ser monitoreados en busca de hiperlipidemia. Rabdomiólisis: en estudios clínicos, fue bien tolerada la administración concomitante de RAPAMUNE* e inhibidores de la HMG CoA reductasa y/o fibratos. Durante la terapia con RAPAMUNE* con o sin CsA, los pacientes deberán ser vigilados en cuanto a los niveles de lípidos, y a los que se les administra un inhibidor de la HMG-CoA reductasa y /o fibratos deberan ser monitoreados por el posible desarrollo de rabdomiólisis y otros efectos adversos, como se describe en la información correspondiente para estos agentes. Función renal: los pacientes tratados con CsA y RAPAMUNE* tuvieron niveles más altos de creatinina sérica y tasas de filtración glomerular más bajas, comparados con pacientes tratados con control de CsA y placebo o aza
Restricciones de uso durante el embarazo y la lactancia: Embarazo: no hay estudios de RAPAMUNE* en mujeres embarazadas. En estudios en animales fue manifiesta, toxicidad embrión/fetal como mortalidad y peso fetal reducido (con retrasos asociados a la osificación del esqueleto). RAPAMUNE* debe ser empleado durante el embarazo solamente si el beneficio potencial sobrepasa el riesgo potencial para el embrión/feto. (Ver Precauciones generales). Uso durante la lactancia: sirolimus es excretado en trazas en la leche de ratas en lactancia. No se sabe si sirolimus es excretado en la leche humana. Debe tomarse una decisión sobre si discontinuar la alimentación al seno materno o discontinuar la terapia con RAPAMUNE*.
Reacciones secundarias y adversas: La frecuencia de las reacciones adversas listadas en la siguiente tabla incluye reacciones reportadas en pacientes tratados con RAPAMUNE* en combinación con CsA y corticosteroides. En general, los eventos adversos relacionados a la administración de RAPAMUNE* fueron dependientes de la dosis / concentración. Las reacciones adversas están listadas en la tabla en las categorías de frecuencia de CIOMS: muy común: ≥10%. Común: ≥1% y < 10%. Poco común: ≥0,1% y < 1%. Raro: ≥0,01% y < 0,1%. Muy raro: < 0,01%. Organismo completo: muy común: linfocele; edema periférico, fiebre, dolor de cabeza, dolor. Común: anormalidades en la cicatrización; edema, infecciones micóticas, virales y bacterianas; (tales como infecciones micobacterianas, incluyendo tuberculosis, virus Epstein-Barr, CMV, y herpes zóster); herpes simple; sepsis. Raro: linfedema. Cardiovascular: muy común: hipertensión. Común: taquicardia, tromboembolismo venoso (incluyendo embolia pulmonar, trombosis venosa profunda). Raro: efusión pericárdica (incluyendo efusiones con impacto hemodinámico significativo en adultos y niños. Digestivo: muy común: dolor abdominal; diarrea, constipación, náusea. Común: estomatitis, ascitis. Poco común: pancreatitis}. Hematológico/linfático: muy común: particularmente a dosis altas: anemia; trombocitopenia. Común: leucopenia; neutropenia; púpura trombocitopénica trombótica/síndrome urémico hemolítico. Poco común: linfoma/ trastorno linfoproliferativo post-trasplante, pancitopenia. Sistema inmune: rara: se han asociado con la administración de sirolimus (ver Advertencias) reacciones de hipersensibilidad, incluyendo reacciones anafilácticas/anafilactoides, angioedema, y vasculitis hipersensible (ver Precauciones generales). Metabólico/nutricional: muy común: hipertrigliceridemia (hiperlipemia); hipercolesterolemia; hipofosfatemia; hiperglicemia; hipokalemia; incremento de deshidrogenasa láctica (LDH); creatinina incrementada. Común: anormalidades en las pruebas de funcionamiento hepático; incremento en TGO y TGP; acumulación de líquidos. Musculoesquelético: muy común: artralgia. Común: necrosis ósea. Respiratorio: común: epistaxis; efusión pleural; neumonía, neumonitis. Poco común: hemorragia pulmonar. Raro: proteinosis alveolar. Piel: muy común: acné. Común: eritema, carcinoma de células escamosas, carcinoma de células basales. Poco común: melanoma. Raro: dermatitis exfoliativa (Ver Advertencias). Urogenital: muy común: infección de vías urinarias. Común: pielonefritis, Proteinuria. Poco común: síndrome nefrótico. Reacciones adversas en las que la frecuencia es desconocida: glomeruloesclerosis segmental focal. RAPAMUNE* después de la discontinuación de CsA: fue determinada la incidencia de reacciones adversas a través de un estudio controlado, multicéntrico, aleatorizado de 60 meses en el cual 215 pacientes con trasplante renal recibieron RAPAMUNE* como régimen de mantenimiento después de la discontinuación de CsA, y 215 pacientes recibieron RAPAMUNE* con terapia CsA. Todos los pacientes fueron tratados con corticosteroides. El perfil de seguridad previo a la aleatorización (inicio de la discontinuación de CsA) fue similar al de los grupos de RAPAMUNE* 2 mg en estudios de RAPAMUNE* en combinación con CsA. Después de la aleatorización (a los tres meses), los pacientes que habían eliminado CsA de su terapia experimentaron incidencias significativamente mayores de incremento de AST/SGOT e incremento de ALT/SGPT, daño hepático, hipocalemia, trombocitopenia, cicatrización anormal, acné, íleo, y trastornos de articulaciones. A la inversa, fue significativamente mayor en pacientes que permanecieron en CsA que en aquellos que se retiraron de la terapia CsA la incidencia de acidosis, hipertensión, toxicidad a CsA, aumento de creatinina, función renal anormal, nefropatía tóxica, edema, hiperuricemia, gota, hiperplasia gingival. La media de la presión sanguínea diastólica y sistólica mejoró significativamente después de la discontinuación de CsA. Después de la discontinuación de CsA, (a los 60 meses), la incidencia de infección por herpes zóster fue significativamente menor en pacientes que recibieron RAPAMUNE* después de la discontinuación de CsA, comparado con pacientes que continuaron recibiendo RAPAMUNE* y CsA. La incidencia de neoplasias malignas después de la discontinuación de CsA, basado en categorías distintas, se presenta en la siguiente tabla. La incidencia de linfoma/enfermedad linfoproliferativa fue similar en todos los grupos de tratamiento. La incidencia total de neoplasias malignas, basado en los pacientes que tuvieron una o más neoplasias fue menor en pacientes que tenían discontinuación de CsA que en pacientes recibiendo RAPAMUNE* más CsA (10,7% versus 15,8%, respectivamente).
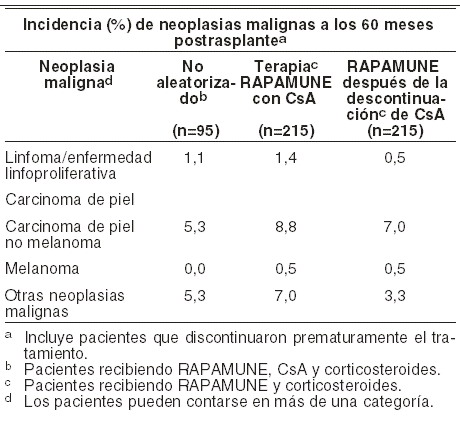
Ya a los 60 meses, la incidencia de neoplasias no de piel (linfoma/enfermedad linfoproliferativa más otras neoplasias de la tabla de arriba), fueron significativamente mayores en el cohorte que continuaron con CsA comparado con el cohorte que tuvo discontinuación de CsA (8,4% vs 3,8%, respectivamente). Para el cáncer de piel, la mediana del tiempo para la primera aparición se retrasó significativamente (491 vs 1.126 días) y cuando se tomó en consideración que el paciente podría tener múltiples cánceres de piel el riesgo relativo (RR = 0,346) para el desarrollo de cáncer de piel disminuyó significativamente en el grupo de discontinuación de CsA comparado con el grupo que continuó con CsA. Se evaluó la seguridad en un estudio controlado (ver Farmacocinética y farmacodinamia) que incluyó 448 pacientes que recibieron una dosis del fármaco del estudio (población de seguridad); 224 pacientes recibieron por lo menos una dosis de sirolimus con tacrolimus, y 224 pacientes recibieron por lo menos una dosis de sirolimus con ciclosporina. En general, la incidencia y naturaleza de los eventos adversos fue similar a los observados en estudios anteriores de combinación con RAPAMUNE*. Diarrea y herpes simple se presentó con mucho más frecuencia en pacientes que recibieron sirolimus y tacrolimus, en tanto que hipertensión, cardiomegalia, linfocele, incremento en la concentración de creatinina, acné, trastorno de vías urinarias, quiste ovárico, y toxicidad asociada con los inhibidores de calcineurina se presentaron en una tasa significativamente más alta en pacientes que recibieron sirolimus y ciclosporina. La incidencia de malignidad fue baja (1,3% en cada grupo). Se valoró la seguridad en un estudio clínico controlado en pacientes pediátricos trasplantados renales ( < 18 años de edad) considerados como de alto riesgo inmunológico, definidos como una historia de uno o más episodios de rechazo agudo y /o la presencia de nefropatía en una biopsia renal (ver Farmacocinética y farmacodinamia). El uso de RAPAMUNE* en combinación con inhibidores de calcineurina y corticosteroides se asoció con un riesgo incrementado de la función renal, anomalías en los lípidos séricos (incluyendo pero no limitado a un incremento de los triglicéridos séricos y colesterol), e infecciones del tracto urinario. No se ha establecido la seguridad y eficacia de la conversión de inhibidores de calcineurina a RAPAMUNE* en el mantenimiento de pacientes con trasplante renal. En un estudio en curso evaluando la eficacia y seguridad de la conversión de inhibidores de calcineurina a RAPAMUNE* (niveles de sirolimus objetivo de 12-20 ng/ml por análisis cromatográfico) en pacientes en terapia de mantenimiento de trasplante renal, se detuvo el enrolamiento en los pacientes del grupo (n=90) con tasa de filtración glomerular basal menor de 40 ml/min. Hubo una mayor tasa de eventos adversos serios, incluyendo neumonía, rechazo agudo, pérdida del injerto y muerte en este brazo de tratamiento con RAPAMUNE* (n=60, mediana de tiempo post-trasplante 36 meses). El uso concomitante de sirolimus con un inhibidor de calcineurina podría incrementar el riesgo de síndrome hemolítico urémico inducido por inhibidor de calcineurina/púrpura trombocitopénica trombótica (microangiopatía trombótica. (ver Precauciones generales). En pacientes con función del injerto demorada, RAPAMUNE* puede demorar la recuperación de la función renal. Enfermedad pulmonar Intersticial: han ocurrido casos de enfermedad pulmonar intersticial (incluyendo neumonitis y raramente bronquiolitis obliterante organizada como neumonía (BOOP) y fibrosis pulmonar) algunos fatales, sin etiología infecciosa identificada, en pacientes recibiendo esquemas de inmunosupresión que incluyen RAPAMUNE*. En algunos casos la enfermedad pulmonar intersticial se ha resuelto una vez que se suspende el tratamiento o se reduce la dosis de RAPAMUNE*. El riesgo puede incrementarse a medida que la concentración mínima de sirolimus aumenta. Infecciones virales latentes: se ha observado el virus BK asociado con neuropatía en pacientes que reciben inmunosupresores, incluyendo RAPAMUNE*. Esta infección puede estar asociada con consecuencias serias, incluyendo la pérdida del injerto. (ver Precauciones generales, Infecciones virales latentes). Hepatotoxicidad: ha sido reportada hepatotoxicidad, incluyendo necrosis hepática fatal con elevadas concentraciones mínimas de sirolimus (ej.: excediendo niveles terapéuticos). Cicatrización anómala: se ha reportado cicatrización anómala después de la cirugía de trasplante, incluyendo dehiscencia fascial, hernia incisional y dehiscencia de anastomosis (ej., herida, vascular, vía aérea, ureteral, biliar). Otras experiencias clínicas: con el uso de RAPAMUNE* se ha reportado azoospermia y ha sido reversible después de la discontinuación del RAPAMUNE* en la mayoría de los casos (ver Datos de seguridad preclínica)
Interacciones medicamentosas y de otro género: 10,1 Inhibidores e inductores del citocromo P450 3A4 (CYP3A4) y P-glicoproteína (P-gp): no se recomienda la coadministración de RAPAMUNE* con inhibidores potentes de CYP3A4 (tales como ketoconazol, voriconazol, itraconazol, telitromicina o claritromicina) o inductores de CYP3A4 (tales como rifampicina o rifabutina). Sirolimus es metabolizado extensamente por la isoenzima CYP3A4 en la pared intestinal y el hígado y experimenta un contra trasporte desde los enterocitos del intestino delgado por la bomba de eflujo de medicamentos de la P-glicoproteína (P-gp). Por lo tanto, la absorción y la eliminación posterior del sirolimus absorbido sistémicamente podría estar influido por los medicamentos que afectan estas proteínas. Los inhibidores de CYP3A4 y P-gp pueden incrementar los niveles de sirolimus. Los inductores de CYP3A4 y P-gp pueden disminuir los niveles de sirolimus. Se deberán de considerar agentes terapéuticos alternativos con menos potencial de inhibición o inducción de CYP3A4 y P-gp en pacientes en los que están indicados los inhibidores o inductores potentes de CYP3A4 y P-gp. Sustancias que inhiben CYP3A4 incluyen pero no limitadas a: bloqueadores de los canales de calcio: diltiazem, nicardipina, verapamil. Agentes antimicóticos: clotrimazol, fluconazol, itraconazol, ketoconazol, voriconazol. Antibióticos: claritromicina, eritromicina, telitromicina, troleandomicina. Agentes procinéticos gastrointestinales: cisaprida, metoclopramida. Otros medicamentos: bromocriptina, cimetidina, CsA, danazol, inhibidores de VIH-proteasa (ej. ritonavir, indinavir). Jugo de toronja. Sustancias que inducen CYP3A4 incluyen pero no limitadas a: anticonvulsivantes: carbamazepina, fenobarbital, fenitoína. Antibióticos: rifabutina, rifampicina, rifapentina. Preparaciones herbolarias: St. John´s Wort (Hypericum perforatum, hipericina). Abajo se discute la interacción farmacocinética entre sirolimus y medicamentos administrados concomitantemente. Se han conducido estudios de interacción con los siguientes medicamentos: diltiazem: diltiazem es un sustrato e inhibidor de CYP3A4 y P-gp. Los niveles de sirolimus deben ser monitoreados y puede ser necesaria la reducción de dosis si se coadministra diltiazem. La administración oral simultánea de 10 mg de solución oral de RAPAMUNE* y 120 mg de diltiazem a 18 voluntarios sanos incrementó significativamente la biodisponibilidad de sirolimus. Los parámetros farmacocinéticos de sirolimus de Cmáx, tmáx y ABC se incrementaron 1,4-, 1,3- y 1,6- veces, respectivamente. Sirolimus no afectó la farmacocinética de diltiazem o sus metabolitos desacetildiltiazem y desmetildiltiazem. Verapamilo: el verapamilo es un inhibidor de CYP3A4. Se deberá de considerar el monitoreo de niveles de RAPAMUNE* y las reducciones adecuadas de las dosis de ambos medicamentos. La administración de múltiples dosis de verapamilo y solución oral de sirolimus afecta significativamente la proporción y el alcance de la absorción de ambos medicamentos. Se incrementaron la Cmáx, tmáx, y ABC en 2,3 veces, 1,1 veces, y 2,2 veces el sirolimus en sangre total respectivamente. Se incrementaron el plasma S-(-) verapamilo Cmáx, y ABC 1,5 veces, y la tmáx, disminuyó un 24%. Eritromicina: la eritromicina es un inhibidor de CYP3A4. Se deberán de monitorear los niveles de RAPAMUNE*; deben considerarse las reducciones adecuadas de la dosis de ambos medicamentos. La administración de múltiples dosis de etilsuccinato de eritromicina y solución oral de RAPAMUNE* aumenta significativamente la proporción y alcance de ambos medicamentos. Se incrementaron la Cmáx, tmáx, y ABC en 4,4 veces, 1,4 veces, y 4,2 veces el RAPAMUNE* en sangre total respectivamente. Ketoconazol: el ketoconazol es un fuerte inhibidor del CYP3A4 y P-gp. No se recomienda la coadministración de ketoconazol y RAPAMUNE*. La administración de dosis múltiples de ketoconazol afecta significativamente la tasa y extensión de la absorción y exposición después de la administración de RAPAMUNE* Solución oral, como se refleja en Cmáx, tmáx y ABC de sirolimus de 4,4, 1,4 y 10,9 veces, respectivamente. Sin embargo, la vida media terminal de sirolimus t ½ no se modificó. Dosis únicas de sirolimus no afectan el estado de equilibrio de las concentraciones plasmáticas de ketoconazol durante 12 horas. Rifampicina: la rifampicina es un fuerte inductor de CYP3A4 y P-gp. No se recomienda la coadministración de rifampicina y RAPAMUNE*. El pretratamiento de 14 voluntarios sanos con dosis múltiples de rifampicina, (600 mg diariamente durante 14 días), seguido por una dosis única de 20 mg de RAPAMUNE* Solución oral, aumentó grandemente la depuración de dosis oral de sirolimus 5,5 veces (rango 2,8 - 10), lo que representa disminuciones medias en ABC y Cmáx de alrededor de 82% y 71%, respectivamente. CsA: la CsA es un sustrato e inhibidor de la CYP3A4 y P-gp. Los pacientes que reciban administración de sirolimus con CsA deben ser monitoreados para el desarrollo de rabdomiólisis (Ver Precauciones generales). Ciclosporina microemulsión [(ciclosporina, USP modificado): se recomienda que RAPAMUNE* sea tomado 4 horas después de la administración de ciclosporina microemulsión. En un estudio de dosis única de interacción entre fármacos, 24 voluntarios sanos recibieron RAPAMUNE* Solución oral 10 mg ya fuera simultáneamente o 4 horas después de una dosis de 300 mg de ciclosporina microemulsión. Para la administración concomitante, los valores medios de Cmáx y ABC aumentaron 116% y 230%, respectivamente, en relación con la administración de RAPAMUNE* como agente único. Sin embargo, cuando fue administrado 4 horas después de la administración de ciclosporina microemulsión, los valores de Cmáx y ABC aumentaron 37% y 80%, respectivamente, comparados con la administración de RAPAMUNE* como agente único. En un estudio idéntico, RAPAMUNE* fue administrado como una dosis de 10 mg en tableta. En la administración simultánea se incrementó, la Cmáx media y el ABC por 6,1 veces y 2,5 veces, respectivamente, relativo a la administración de RAPAMUNE* solo. Sin embargo, cuando se administró 4 horas después de la administración de ciclosporina microemulsión, incrementaron la Cmáx y ABC de sirolimus por sólo un 33% comparado con la administración de RAPAMUNE* solo. Luego de la administración de dosis múltiples de RAPAMUNE* Solución oral administrado 4 horas después de ciclosporina en pacientes post-trasplante renal a lo largo de 6 meses, la depuración de dosis oral de ciclosporina estuvo reducida, y se necesitaron dosis menores de CsA microemulsión (Neoral®) para mantener la concentración blanco de ciclosporina. RAPAMUNE* Solución oral: en un estudio de interacción medicamento-medicamento, se les administró a 24 sujetos voluntarios 5 mg de RAPAMUNE*, cualquiera de las dos opciones simultáneamente o dos horas antes y después de una dosis de 300 mg de ciclosporina microemulsión. En la administración simultánea, la media Cmáx, y ABC de sirolimus se incrementaron un 117% y 183%, respectivamente, relativo a la administración de RAPAMUNE* solo. Cuando se administró 2 horas después de la administración de ciclosporina microemulsión, la Cmáx, de sirolimus y el ABC se incrementaron un 126% y 141%, respectivamente, comparado con la administración de RAPAMUNE* solo. Cuando se administraron 2 horas antes de la administración de ciclosporina microemulsión, no se afectaron la Cmáx, y ABC de sirolimus. CsA Solución oral: en un estudio de dosis múltiples con 150 pacientes con psoriasis, RAPAMUNE* Solución oral 0,5, 1,5, y 3 mg /m2 /día fue administrado simultáneamente con CsA solución oral 1.25 mg /kg /día. El aumento en las concentraciones mínimas promedio de RAPAMUNE* estuvo entre 67% - 86% con respecto a cuando el RAPAMUNE* fue administrado sin CsA. No hubo ningún efecto significativo de dosis múltiples de sirolimus sobre las concentraciones mínimas de Sandimmune® (ciclosporina, solución oral, USP). Sin embargo, el CV% fue más alto (entre 85,9% - 165%) que el de los estudios previos. Inhibidores de HMG-CoA reductasa, Fibratos: a los pacientes que se les administra RAPAMUNE* con inhibidores de HMG-CoA reductasa y/o fibratos se les debe monitorear por el desarrollo de rabdomiólisis. Inhibidores calcineurina: se ha reportado en pacientes recibiendo sirolimus con un inhibidor de calcineurina, síndrome hemolítico urémico inducido por inhibidor de calcineurina/púrpura trombocitopénica trombótica (microangiopatía trombótica). Vacunación: los inmunosupresores pueden afectar la respuesta a la vacunación. Durante el tratamiento con inmunosupresores, incluyendo RAPAMUNE*, la vacunación puede ser menos efectiva. Debe evitarse el uso de vacunas vivas durante el tratamiento con RAPAMUNE*. Ejemplos de vacunas vivas son: las de sarampión, parotiditis, rubéola, polio oral, BCG, fiebre amarilla, varicela, y TY21a tifoide. Fármacos que pueden ser coadministrados sin ajustes a la dosis: no se observaron interacciones farmacocinéticas clínicamente significativas en los estudios con los siguientes fármacos: aciclovir, atorvastatina, digoxina, glibenclamida (gliburida), nifedipina, norgestrel 0,3 mg/ etinil estradiol 0,03 mg, metilprednisolona, y trimetoprim/sulfametoxazol. Interacción con alimentos: la biodisponibilidad de RAPAMUNE* se afecta por ingestión concomitante de alimentos después de la administración ya sea de la solución oral o las grageas. RAPAMUNE* debe ser administrado consistentemente con o sin alimentos para minimizar la variabilidad de los niveles sanguíneos. El jugo de toronja reduce el metabolismo del medicamento mediado por CYP3A4 y potencialmente incrementa el contra trasporte desde los enterocitos del intestino delgado mediado por P-gp. Este jugo no debe ser tomado con solución oral o grageas de RAPAMUNE* o utilizado para la dilución de la solución oral (Ver Dosis y vía de administración).
Alteraciones en los resultados de pruebas de laboratorio: No hay estudios sobre las interacciones de sirolimus con pruebas de laboratorio clínico comúnmente empleadas.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Carcinogenicidad: se realizaron estudios de carcinogénesis en ratones y ratas. En un estudio en ratones hembra a 86 semanas, a 4 dosificaciones, que fueron aproximadamente de 16 a 135 veces las dosis clínicas (ajustadas para superficie corporal) hubo un aumento estadísticamente significativo en linfomas malignos a todos los niveles de dosis, comparados con los de los controles. En un segundo estudio en ratones a dosis que fueron aproximadamente de 3 a 16 veces las dosis clínicas (ajustadas para la superficie corporal), se consideraron relacionadas con el sirolimus el adenoma hepatocelular y el carcinoma (ratones machos). En el estudio en rata a 104 semanas, a dosificaciones que fueron aproximadamente 0,4 a 1 veces las dosis clínicas (ajustadas para la superficie corporal), hubo un aumento estadísticamente significativo en la incidencia de adenoma testicular en el grupo de dosis mayores. Mutagenicidad: sirolimus no fue genotóxico en el ensayo in vitro de mutación bacteriana reversa, el ensayo de aberraciones cromosómicas en células de ovario de hámster chino, el ensayo de mutación prospectivo en células de linfoma de ratón, o in vivo en el ensayo de micronúcleos en ratón. Toxicología reproductiva: sirolimus fue tóxico embrio/fetal en ratas a dosis de 0,1 mg/kg y más (aproximadamente 0,2 a 0,5 las dosis clínicas ajustadas por área de superficie corporal). La toxicidad embrio/fetal se manifestó como mortalidad y peso fetal reducido (con retrasos asociados en la osificación esquelética). Sin embargo, no fue evidente teratogénesis. Las ratas incrementaron la mortalidad embrio/fetal en combinación con CsA, comparado con sirolimus solo. No hubo efectos en el desarrollo de los conejos a la dosis materna tóxica de 0,05 mg/kg (aproximadamente 0,3 a 0,8 veces la dosis clínica ajustada por área de superficie corporal). No hubo ningún efecto sobre la fertilidad en ratas hembra después de la administración de sirolimus a dosificaciones de hasta 0,5 mg /kg (aproximadamente 1 a 3 veces las dosis clínicas ajustadas por área de superficie corporal). En ratas macho, hubo una ligera reducción en la fertilidad, comparada con los controles en un estudio, a una dosificación de 2 mg /kg (aproximadamente 4 a 11 veces las dosis clínicas ajustadas por área de superficie corporal). Un segundo estudio falló para confirmar este hallazgo. Se observó las reducciones en los pesos testiculares y/o lesiones histológicas (ej., atrofia tubular y células gigantes tubulares) en ratas después de dosis de 0,65 mg/kg (aproximadamente 1 a 3 veces las dosis clínicas ajustadas para la superficie corporal) y más en un estudio en monos a 0,1 mg/kg (aproximadamente 0,4 a 1 veces las dosis clínicas ajustadas para la superficie corporal) y más. Las cuentas en esperma fueron reducidas en ratas macho después de la administración de sirolimus durante 13 semanas a una dosis de 6 mg/kg (aproximadamente 12 a 32 veces las dosis clínicas ajustadas para la superficie corporal), pero mostraron una mejoría después de 3 meses de la suspensión de la dosis.
Dosis y vía de administración: Dosis: no se ha determinado la biodisponibilidad de las grageas después de que se aplastan, mastican o dividen; por lo tanto, esto no se puede recomendar. A los pacientes incapaces de ingerir las grageas se les deberá de prescribir la solución e instruirlos en su uso. Se ha demostrado que dos miligramos (2 mg) de RAPAMUNE* Solución oral son clínicamente equivalentes a 2 mg de RAPAMUNE* Grageas; por lo tanto, son intercambiables en una base de mg a mg. Sin embargo, se desconoce si las dosis mayores de RAPAMUNE* Solución oral son equivalentes clínicamente a dosis mayores de RAPAMUNE* Grageas en una base mg a mg. Deberán prescribir RAPAMUNE* sólo médicos con experiencia en terapia inmunosupresora y manejo de pacientes con trasplante de órganos. Los pacientes que reciben el medicamento deben ser manejados en instalaciones equipadas y personal con recursos adecuados de laboratorio y de soporte médico. El médico responsable de la terapia de mantenimiento deberá de tener los requisitos completos de información para el seguimiento del paciente. Pacientes con riesgo inmunológico bajo a moderado: terapia de combinación RAPAMUNE* y CsA: para los receptores de trasplantes de novo, se deberá de administrar una dosis de carga de RAPAMUNE* correspondiente a tres veces la dosis de mantenimiento. Se recomienda una dosis de mantenimiento diaria de 2 mg para el uso en pacientes de trasplante renal, con una dosis de carga de 6 mg. A pesar que fue utilizada en los estudios clínicos una dosis de mantenimiento diaria de 5 mg, con una dosis de carga de 15 mg, de la solución oral y mostró ser segura y efectiva, no se pudo establecer una ventaja en la eficacia sobre la dosis de 2 mg para los pacientes de trasplante renal. Los pacientes que recibieron por día 2 mg de RAPAMUNE* Solución oral demostraron un mejor perfil de seguridad total que los pacientes que recibieron 5 mg de RAPAMUNE* Solución oral por día. Se recomienda que la solución oral y grageas de RAPAMUNE* sean utilizados en un régimen con CsA y corticosteroides, la ciclosporina deberá ser retirada 2 a 4 meses después del trasplante renal en pacientes con un riesgo inmunológico bajo a moderado y se debe aumentar la dosis de RAPAMUNE* hasta llegar a las concentraciones sanguíneas recomendadas. No se ha estudiado la suspensión de ciclosporina en pacientes con rechazo agudo grado III de Banff 93 o rechazo vascular antes de suspender CsA, en pacientes dependientes de diálisis o con creatinina sérica > 4,5 mg/dl, pacientes de raza negra, retransplantes, transplantes de múltiples órganos o pacientes con panel de títulos altos de anticuerpos reactivos (ver Indicaciones y Farmacocinética y farmacodinamia). RAPAMUNE* después de la discontinuación de CsA (referido como el régimen de mantenimiento de RAPAMUNE* RMR): inicialmente, los pacientes deberán estar recibiendo una terapia combinada de RAPAMUNE* y CsA. De los 2 a 4 meses después del trasplante, la CsA deberá ser discontinuada progresivamente durante 4 a 8 semanas y la dosis de RAPAMUNE* deberá ser ajustada para obtener concentraciones de sangre total dentro de un rango de 16 a 24 ng/ml (método cromatográfico) durante el primer año después del trasplante. De ahí en adelante, las concentraciones objetivo deberán ser de 12 a 20 ng/ml (método cromatográfico). Las observaciones actuales al año uno y 5 (ver abajo) fueron cercanas a estos rangos (véase abajo monitorización sanguínea de la concentración de sirolimus). El monitoreo terapéutico del fármaco no deberá de ser solamente la base para el ajuste de la terapia de RAPAMUNE*. Se deberá tener atención cuidadosa a los síntomas/signos clínicos, biopsias titulares, y parámetros de laboratorio, la ciclosporina inhibe el metabolismo y trasporte de sirolimus, y consecuentemente, las concentraciones de sirolimus disminuirán cuando la CsA es discontinuada, a menos que la dosis de RAPAMUNE* sea incrementada. La dosis de RAPAMUNE* necesitará ser aproximadamente 4 veces mayor para considerar tanto la ausencia de interacción farmacocinética (aproximadamente aumento de dos veces) como el requerimiento aumentado inmunosupresor en ausencia de CsA (aproximadamente aumento de dos veces). La dosis de RAPAMUNE* necesitará ser aproximadamente 4 veces mayor para contar para ambos la ausencia de interacción farmacocinética
Descripción.
Metodología de análisis: los rangos de concentración mínima de sirolimus recomendados en 24 horas están basados en métodos cromatográficos. Se han utilizado varias metodologías analíticas para medir la concentración de sirolimus en sangre total. Actualmente en la práctica clínica, las concentraciones de sirolimus en sangre total son medidas por ambas metodologías cromatografía e inmunoensayo. Los valores de concentración obtenidos por estas metodologías diferentes no son intercambiables. Se deberán hacer ajustes al rango objetivo de acuerdo con el análisis utilizado para determinar la mínima concentración de sirolimus. Ya que los resultados son dependientes del análisis de laboratorio, y los resultados pueden cambiar con el tiempo, se deberá de realizar un ajuste al rango terapéutico objetivo con conocimiento detallado de los análisis específicos en el sitio. Modo de administración: RAPAMUNE* está propuesto solamente para la administración oral. RAPAMUNE* debe ser ingerido consistentemente ya sea con los alimentos o sin ellos para minimizar la variación en la absorción del fármaco. Para la dilución de RAPAMUNE* solución oral deberá ser usada solamente agua o jugo de naranja, utilizando solamente vasos de vidrio o de plástico. No diluir RAPAMUNE* Solución oral con jugo de toronja (véase interacciones medicamentosas) o con otros líquidos. RAPAMUNE* Solución oral contiene polisorbato-80, que se sabe incremente la tasa de extracción de di-(2-etilhexil)ftalato (DEHP) del cloruro de polivinilo (PVC). Esto debe considerarse durante la preparación y administración de solución oral de RAPAMUNE*. Es importante que las recomendaciones de dosis se sigan estrechamente. Instrucciones para la dilución y administración de RAPAMUNE* Solución oral: frascos: antes de la administración debe utilizarse para la dilución de RAPAMUNE* Solución oral un recipiente de vidrio o plástico. La jeringa ámbar de dosificación oral debe ser usada para retirar la cantidad prescrita de RAPAMUNE* ® Solución oral del frasco. Vacíe la cantidad correcta de RAPAMUNE* de la jeringa solamente en un recipiente de vidrio o plástico que tenga cuando menos 1/4 de taza, (60 ml ó 2 onzas) de agua o jugo de naranja. No deben usarse otros líquidos, incluyendo jugo de toronja, para la dilución. Agite vigorosamente y beba de inmediato. Vuelva a llenar el recipiente de vidrio con un volumen adicional (mínimo de ½ taza, (120 ml ó 4 onzas) de agua o jugo de naranja, agite vigorosamente, y beba de inmediato. Manejo y desecho: dado que RAPAMUNE* no se absorbe a través de la piel, no hay precauciones especiales. Sin embargo, si ocurre contacto directo con la piel o membranas mucosas, lave cuidadosamente con jabón y agua; enjuague los ojos con agua simple.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: La experiencia con sobredosis es limitada. En general, los efectos adversos de una sobredosis con sirolimus son consistentes con aquellos listados en la sección de reacciones adversas. Deben tomarse medidas de apoyo generales en todos los casos de sobredosis. Con base en la pobre solubilidad acuosa y la alta unión a eritrocitos y proteínas plasmáticas de sirolimus, se anticipa que RAPAMUNE* no sea dializable en ningún grado significativo. En ratones y ratas, la DL50 oral aguda fue mayor de 800 mg /kg.
Presentación(es): RAPAMUNE* Solución oral tiene una concentración de 1 mg /ml en: caja con un frasco con 60 ml, 30 jeringas dosificadoras y estuche de viaje. RAPAMUNE* Grageas de 1 mg y 2 mg. Caja con 30 grageas. Caja con 60 grageas.
Recomendaciones sobre almacenamiento: Los frascos de RAPAMUNE* ® Solución oral deben ser almacenados protegidos de la luz y refrigerados a entre 2C y 8C (36F y 46F). Una vez abierto el frasco, su contenido debe ser usado dentro de un plazo de un mes. Si es necesario, el paciente puede almacenar los frascos a temperatura ambiente de hasta 25°C (77°F) por un período de tiempo corto (por ej., varios días, pero no más de 30 días). Se proporciona una jeringa ámbar y tapa para la dosificación, y el producto puede ser mantenido en la jeringa por un máximo de 24 horas a temperatura ambiente de hasta 25C (77F) o refrigerado a 2C a 8C (36F a 46F). La jeringa debe desecharse después de un sólo uso. Después de la dilución, la preparación debe ser usada inmediatamente. RAPAMUNE* Solución oral en frascos puede desarrollar un enturbiamiento ligero cuando se refrigera; este enturbiamiento no tiene efecto en la calidad del producto. Si tal enturbiamiento ocurre permita que el producto adquiera la temperatura ambiente, y agite suavemente hasta que el enturbiamiento desaparezca. RAPAMUNE* Grageas deberá ser almacenado a no más de 25°C. Use las cajas para proteger los envases de burbuja de la luz.
Leyendas de protección: No se deje al alcance de los niños. Su venta requiere receta médica. Consérvese en refrigeración entre 2° y 8°C. Grageas: consérvese a temperatura ambiente a no más de 25°C. Protéjase de la luz. No se use en el embarazo. Literatura exclusiva para médicos.
Nombre y domicilio del laboratorio: Wyeth, S.A. de C.V. Poniente 134 No. 740, Col. Industrial Vallejo, 02300, México, D.F. *Marca Registrada.
Número de registro del medicamento: 099M2000 SSA IV y 125M2001 SSA IV
Clave de IPPA: 103300203A0374


 Cambiar de país
Cambiar de país