ALVERITIN
ALVARTIS PHARMA
Denominación genérica: Epoetin alfa. Eritropoyetina humana recombinante (r-Hu-EPO).
Farmacocinética y farmacodinamia: Solución inyectable. Cada frasco ámpula con solución contiene: eritropoyetina humana recombinante alfa 1.000 UI, 4.000 UI. Vehículo cbp 1 ml.
Indicaciones terapéuticas: Anemia de la insuficiencia renal crónica.
Farmacocinética y farmacodinamia: Las células endoteliales de la corteza renal y de la médula externa son, al parecer, las responsables de la síntesis de eritropoyetina (Lacombe, 1988). Después de la síntesis en el riñón y su liberación a la sangre, la eritropoyetina desarrolla su acción hormonal en la médula ósea estimulando el desarrollo de las células precursoras eritroides, en eritrocitos maduros, estimula no sólo la multiplicación de las células precursoras eritropoyéticas sino también su diferenciación. En el estadio de maduración, tiene lugar un aumento de la hemoglobinización y una aceleración de la maduración de los eritroblastos, así como un aumento de los reticulocitos, precursores directos de los eritrocitos. A nivel celular, actúa como una hormona polipeptídica sobre los receptores de las células precursoras eritroides; puede, además, actuar a través de un segundo mensajero e internalizarse. Diferentes grupos, entre ellos Millar (1988), han estudiado la acción bioquímica de la eritropoyetina sobre su ligando a nivel celular. Se ha reportado que puede incrementar notablemente los niveles de calcio intracelular. Vía intravenosa: las investigaciones llevadas a cabo con la eritropoyetina subsiguientemente a la administración de múltiples dosis intravenosas revelaron una vida media de aproximadamente 4 horas en voluntarios y una vida media algo más prolongada en pacientes con insuficiencia renal, aproximadamente 5 horas. Por el momento, se informa una vida media en niños de 6 horas. Vía subcutánea: a continuación de la inyección subcutánea, los niveles séricos son mucho más bajos que los niveles logrados a continuación de la inyección IV; los niveles incrementan lentamente y alcanzan un pico entre las 8 y 12 horas después de la administración. El pico es siempre más bajo que el pico alcanzado empleando la vía IV.
Contraindicaciones: La eritropoyetina se encuentra contraindicada en: 1. Pacientes que padecen de hipertensión no controlada. 2. Pacientes que presenten hipersensibilidad a productos derivados de células superiores. 3. Pacientes que presenten hipersensibilidad a la albúmina humana. 4. Hipersensibilidad al principio activo. 5. Hiperglobulina.
Precauciones generales: Pacientes con enfermedad renal crónica. Hipertensión: en los pacientes con enfermedad renal, es muy importante el control de la hipertensión arterial, ya que más del 80% de estos pacientes tienen una historia anterior de hipertensión; aunque este producto no tiene efecto directo sobre la presión sanguínea, se puede observar un incremento. Durante la primera fase del tratamiento, el 25% aproximadamente de los pacientes pueden requerir del inicio o del incremento en la terapia antihipertensiva. La encefalopatía por hipertensión y convulsiones puede ser observada en pacientes con insuficiencia renal crónica en tratamiento con eritropoyetina. En general, se deben tomar cuidados especiales y chequear o monitorear muy de cerca la presión arterial de los pacientes bajo tratamiento con eritropoyetina. Los pacientes deben ser alertados y deben ser informados de la importancia de seguir rigurosamente el tratamiento antihipertensivo y de la dieta indicada. Si la presión arterial es difícil de controlar con las medidas apropiadas, se debe disminuir el nivel de hematocrito disminuyendo o manteniendo la dosis de eritropoyetina, aunque esta disminución en hematocrito puede demorarse semanas. Es recomendable que la dosis de eritropoyetina se disminuya si el nivel de incremento del hematocrito excede en 4 puntos en cualquier período de 2 semanas, por la posible asociación del incremento excesivo del hematocrito con la exacerbación de la hipertensión. Convulsiones: se ha presentado convulsiones en pacientes que han participado en diferentes ensayos clínicos con eritropoyetina. En pacientes en diálisis, ha habido una incidencia más alta durante los primeros 90 días del tratamiento (aproximadamente un 2,5%). En los primeros 90 días de tratamiento, la presión arterial y el "aura" o signos neurológicos premonitorios deben ser monitoreados muy cercanamente. Los pacientes deben evitar actividades riesgosas como conducir, operar maquinarias, etc., durante este período. Como la relación entre las convulsiones y los niveles de hematocrito es incierta, se recomienda que se disminuya la dosis de eritropoyetina si el hematocrito ha incrementado en 4 puntos en un período cualquiera de 2 semanas. Eventos trombóticos: durante la hemodiálisis, los pacientes en tratamiento con eritropoyetina pueden requerir un incremento de anticoagulantes para prevención del sistema de coagulación del riñón artificial. No se ha relacionado estadísticamente el incremento en Ht y la incidencia de eventos trombóticos (incluyendo trombosis de acceso vascular). En los ensayos clínicos, la incidencia anual de trombo de acceso vascular ha sido de 0,25 por paciente/año, incidencia que es mayor que en los pacientes no tratados que se someten a tratamiento dialítico. En general, los pacientes con insuficiencia renal crónica, en diálisis o no, han tenido una incidencia de otros eventos trombóticos (infarto del miocardio, accidente cerebrovasculares o ataques isquémicos transitorios) en menos de 0,04 eventos por paciente/año de tratamiento con eritropoyetina. Los pacientes con historia de accidentes vasculares deben ser monitoreados cuidadosamente. Pacientes con SIDA en régimen terapéutico con zidovudina: no se ha observado ningún episodio de incremento de la presión arterial, ni de convulsiones ni de eventos trombóticos en estos pacientes. Hematología: se ha observado muy raramente en algunos de los pacientes tratados con eritropoyetina una exacerbación de porfiria; sin embargo, no se ha podido comprobar el incremento de excreción de metabolitos de las porfirinas en voluntarios sanos. No obstante, la eritropoyetina debe ser utilizada cautelosamente en pacientes con porfiria. Respuesta disminuida o retardada: si el paciente falla en responder o mantener la respuesta a dosis en el rango recomendado, esto puede deberse a las siguientes etiologías: 1. Deficiencia de hierro. Virtualmente, todos los pacientes requieren un suplemento de hierro. 2. Infecciones subyacentes, procesos inflamatorios o procesos malignos. 3. Pérdida de sangre oculta. 4. Enfermedades hematológicas subyacentes, por ej., talasemia, anemia refractaria u otro síndrome mielodisplásico. 5. Deficiencias de vitaminas, ácido fólico. 6. Hemólisis. 7. Intoxicación por aluminio. 8. Osteítis por fibrosis quística.
Restricciones de uso durante el embarazo y la lactancia: La eritropoyetina debe ser administrada durante los períodos de embarazo y lactancia sólo en caso claramente necesario y si el potencial beneficio justifica el potencial de riesgo para el feto. No se conoce si la eritropoyetina puede causar perjuicios fetales al ser administrada a mujeres embarazadas o bien, si puede afectar la capacidad reproductora.
Reacciones secundarias y adversas: Los efectos secundarios más importantes desde el punto de vista clínico en los pacientes con insuficiencia renal crónica son la hipertensión, convulsiones, encefalopatía, ataques isquémicos transitorios, trombosis del acceso de la hemodiálisis. Los pacientes con SIDA y de oncología con quimioterapia presentan un cuadro gripal con alguna sintomatología febril y jaqueca. Pacientes con insuficiencia renal crónica: estudios realizados hasta la fecha indican que la eritropoyetina es un producto muy bien tolerado. Las reacciones adversas reportadas son frecuentemente secuelas de la insuficiencia renal crónica y no necesariamente atribuibles a la eritropoyetina. En un ensayo doble ciego placebo controlado con 300 pacientes con insuficiencia renal crónica, los eventos reportados en más del 5% en el grupo tratado fueron: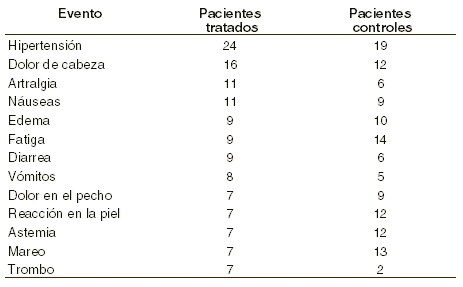
En el ensayo clínico realizado en los Estados Unidos con la eritropoyetina en 567 pacientes en diálisis, la incidencia (número de eventos por pacientes/año) de las reacciones adversas más frecuente fueron la hipertensión, 0,75; dolor de cabeza, 0,40; taquicardia; náuseas y vómito, 0,26; acceso vascular 0,25; falta de aire, 0,14 y diarreas, 0,11. Hipertensión: el incremento en la presión arterial ha sido reportado en los diferentes ensayos clínicos frecuentemente durante los primeros 90 días del tratamiento. En los resultados del ensayo clínico fase II con eritropoyetina, se observó una tendencia a que ocurriera la hipertensión arterial en aquellos pacientes que incrementaban rápidamente su hematocrito a más de 4 puntos; sin embargo, en el ensayo doble ciego se encontraron resultados semejantes entre los dos grupos estudiados, el control y el tratado con EPO. Convulsiones: se han reportado 47 crisis convulsivas en 1.010 pacientes estudiados en diálisis y tratados con eritropoyetina; aunque esta cifra es realmente baja, parece haber una tendencia a que se incremente el riesgo de convulsiones en los primeros 90 días del tratamiento. Eventos trombóticos: durante la hemodiálisis, los pacientes en tratamiento con eritropoyetina pueden requerir un incremento de anticoagulantes para la prevención del sistema de coagulación de riñón artificial. No se ha relacionado estadísticamente el incremento en Ht y la incidencia de eventos trombóticos (incluyendo trombosis de acceso vascular). En los ensayos clínicos, la incidencia anual de trombo de acceso vascular ha sido de 0,25 por paciente/año, incidencia que es mayor que en los pacientes no tratados que se someten a tratamiento dialítico. En 125.000 pacientes tratados con eritropoyetina, se han reportado muy raramente eventos serios de tromboembolismo, incluyendo tromboflebitis migratoria, trombosis microvascular émbolo-pulmonar y trombosis de la arteria de la retina. En general, la frecuencia ha sido de 0,0001 eventos por paciente/año. Reacciones alérgicas: ninguna reacción alérgica ha sido reportada, ni reacción anafiláctica, durante la administración de la eritropoyetina. En 125.000 pacientes tratados con eritropoyetina, no se ha observado ninguna reacción alérgica seria. Ha sido reportada como muy rara urticaria, sola o asociada, 0,0001 eventos por paciente/año. Debe advertirse que, aunque sea un evento muy raro, puede ocurrir. No hay evidencias de anticuerpos desarrollados contra la molécula en pacientes estudiados hasta la fecha, incluyendo pacientes recibiendo eritropoyetina comercial durante 4 años. Pacientes con SIDA en tratamiento con zidovudina: en el ensayo clínico realizado con el eritropoyetina comercial (297 pacientes), se encontraron diversas reacciones adversas. A continuación, se reportan las que tuvieron frecuencia mayor del 10%: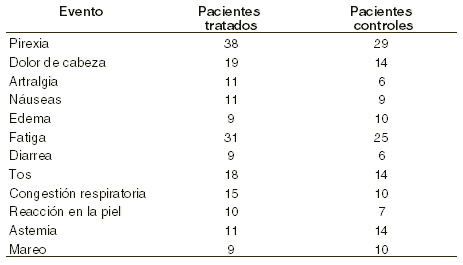
Pacientes con cáncer en tratamiento con quimioterapia: en un estudio clínico doble ciego con placebo en 131 pacientes tratados con la eritropoyetina, se encontraron las siguientes reacciones adversas: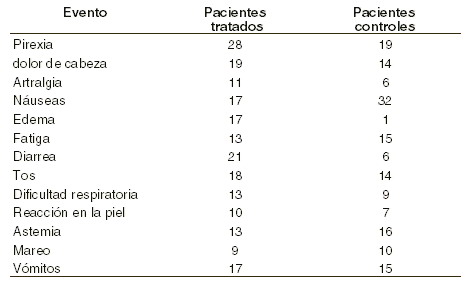
Aunque se observaron algunas diferencias significativas entre el grupo tratado y el de control, se considera que el perfil de seguridad del producto es consistente con el proceso de una enfermedad neoplásica avanzada.
Interacciones medicamentosas y de otro género: No existen evidencias que indiquen que el tratamiento con r-HuEPO altere el metabolismo de otras drogas. Sin embargo, el hecho de que la ciclosporina se una a los glóbulos rojos representa un potencial de interacción de droga. Si la r-HuEPO se administra concomitantemente con ciclosporina, los niveles sanguíneos de ciclosporina deben ser vigilados y la dosis de ciclosporina debe ser adecuada en relación con el incremento del hematocrito. No administrar por infusión intravenosa o conjuntamente con otras soluciones de medicamentos.
Alteraciones en los resultados de pruebas de laboratorio: Las correcciones de la anemia en los pacientes en prediálisis puede dar lugar a un aumento del apetito y de la ingesta de potasio, lo que puede ocasionar hipercalcemia. También, la corrección de la anemia de los pacientes en diálisis puede dar lugar a un incremento de potasio sérico, creatinina y fósforo inorgánico. Estas alteraciones en la química sanguínea deberán manejarse con modificaciones en la dieta o en la diálisis, si ello fuera apropiado.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Carcinogenicidad: no se han llevado a cabo estudios de carcinogenicidad. Mutagenicidad: la r-HuEPO no ha demostrado ningún cambio en el test de mutagenicidad y en el test de micronúcleo. Teratogenicidad: hasta ahora, no existen evidencias de efectos teratogénicos.
Dosis y vía de administración: Endovenosa o intravenosa, subcutánea. Pacientes con insuficiencia renal crónica: dosis de inicio: pueden encontrarse en el rango de 50/100 U/kg tres veces por semana. Esta dosis ha demostrado ser efectiva y segura para incrementar los niveles de hematocrito y eliminar la dependencia y necesidad de transfusiones. La dosis de la eritropoyetina debe ser disminuida cuando el nivel de hematocrito alcanza el 36%. La eritropoyetina puede ser administrada por vía intravenosa o subcutánea. En pacientes en hemodiálisis, puede ser administrada en bolo intravenoso. La administración debe ser independiente de la diálisis, pero puede ser administrada en la vena, al final del procedimiento de diálisis. Ajuste de dosis: a continuación del tratamiento con la eritropoyetina, se debe esperar un período de tiempo de 2 a 4 semanas para que las células progenitoras eritroides maduren y sean liberadas a la circulación y que se produzca finalmente la elevación de los niveles de hematocrito. El ajuste no debe realizarse por más de una vez en un mes, si no es clínicamente imprescindible. Si el nivel de hematocrito se eleva alcanzando 36%, la dosis se debe reducir para mantenerse en ese rango; si no se logra mantener, se recomienda suspender el tratamiento hasta que se disminuya el Ht a esa cifra. Si el nivel de hematocrito se incrementa en cualquier período de dos semanas de tiempo en más de 4 puntos, se debe disminuir la dosis; después de esta reducción, se deben monitorear los niveles de Ht dos veces por semana durante 2 a 6 semanas y después, aplicar la dosis de mantenimiento. Si el nivel de hematocrito no se incrementa en 5/6 puntos en el período de 8 semanas, la dosis debe ser incrementada y evaluada de nuevo pasadas 2-4 semanas, y de nuevo se puede volver a incrementar pasados intervalos de 4-6 semanas. Dosis de mantenimiento: aunque la dosis de mantenimiento se debe individualizar, un promedio de dosis de mantenimiento puede ser de 75 U/kg para pacientes en diálisis, tres veces por semana. Respuesta disminuida o retardada: cerca del 95% de los pacientes con insuficiencia renal crónica que han sido tratados con eritropoyetina han respondido al tratamiento. Si se encuentra algún paciente que no responde, se debe analizar el nivel de reserva de hierro o debe analizarse otra posible etiología de la anemia. Pacientes con SIDA en tratamiento con zidovudina: se recomienda evaluar previamente nivel de eritropoyetina endógena, porque está descrito que, si tiene niveles endógenos de eritropoyetina mayores de 500 U, no responde al tratamiento. Dosis de inicio: 100 U/kg, tres veces por semana durante 8 semanas, si el nivel de eritropoyetina es menor a 500 U y el de zidovudina menor a 4.200 mg semana. Incremento de dosis: si la respuesta no es satisfactoria en reducir transfusiones o elevar Ht en 8 semanas la dosis se puede incrementar en 50 o 100 U tres veces por semana, y la respuesta, evaluarla en 4-8 semanas. Si es necesario de nuevo incrementar de 50-100 sólo hasta 300 U/kg, dosis más altas no son recomendables. Dosis de mantenimiento: después de alcanzar el nivel requerido, se debe mantener de acuerdo con los factores del tratamiento con zidovudina y las infecciones intercurrentes. Tratamiento en pacientes con cáncer en régimen de quimioterapia: se recomienda evaluar previamente el nivel de eritropoyetina endógena, porque está descrito que si tiene niveles endógenos de eritropoyetina mayor de 200 U/ml, no responde al tratamiento. Dosis de inicio: la dosis recomendada es de 150 U/kg tres veces por semana. Incremento de dosis: si la respuesta no es satisfactoria en reducir transfusiones o elevar Ht en 8 semanas, la dosis se puede incrementar en 50 o 100 U tres veces por semana y la respuesta, evaluarla en 4-8 semanas. Si es necesario de nuevo incrementar de 50/100 sólo hasta 300 U/kg, dosis más altas no son recomendables. En general: si la hemoglobina excede los 13 g/dl, la dosificación debe ser interrumpida hasta alcanzar una concentración de hemoglobina de 12 g/dl. Al restituirse el tratamiento, la dosis debe reducirse en un 25% y, subsiguientemente, ajustada para mantener este nivel de hemoglobina. Cuando el tratamiento es suspendido, la concentración de hemoglobina desciente alrededor de 0,5 g/dl por semana.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: La respuesta a la eritropoyetina se relaciona con la dosis y es individual. En el caso de una respuesta hematopoyética excesiva por una sobredosis de eritropoyetina, se debe suspender su administración y puede considerarse una flebotomía (sangría). Ante la presencia de fenómenos hipertensivos o convulsivos que puedan estar relacionados con sobredosis de eritropoyetina, deberá administrarse manejo de sostén.
Presentación(es): ALVERITIN 2000 Ul/ml: caja con 12 y 6 frascos ámpula con 1 ml para venta al público y para venta al sector salud con clave 5332. ALVERITIN 4000 Ul/ml: caja con 6 y 1 frascos ámpula con 1 ml para venta al público y para venta al sector salud con clave 5333.
Recomendaciones sobre almacenamiento: Consérvese a temperatura de 2°C a 8°C, no congelarse ni agitarse. Protéjase de la luz.
Leyendas de protección: Su venta requiere receta médica. No se deje al alcance de los niños. Literatura exclusiva para médicos. Consérvese en refrigeración entre 2 a 8°C, no congelarse ni agitarse. Protéjase de la luz.
Nombre y domicilio del laboratorio: Alvartis Pharma, S.A. de C.V. Carretera Jaltepec- Cd. Sahagún No. 1, San Miguel Jaltepec, Edo. de Méx. C.P. 55965.
Bibliografía: Investigación pura del CIMAB S.A. La Habana, Cuba. Cuadro Básico de Medicamentos.
Número de registro del medicamento: 387M2008 SSA.
Clave de IPPA: KEAR-083300CT051130/R2008