AMITIZA
TAKEDA
Denominación genérica: Lubiprostona
Forma farmacéutica y formulación: Cápsulas. Cada capsula contiene: Lubiprostona 8 mcg y 24 mcg. Excipiente cbp 1 cápsula. Cápsula de 8 mcg: Cápsula de gelatina suave ovalada, rosa con "SPI" impreso de un lado.Cápsula de 24 mcg: Cápsula de gelatina suave ovalada, naranja con "SPI" impreso de un lado.
Indicaciones terapéuticas: Estreñimiento Crónico Idiopático (ECI): AMITIZA está indicado para el tratamiento del estreñimiento crónico idiopático en adultos.Síndrome de Intestino Irritable con Estreñimiento (SII-E): AMITIZA está indicada para el tratamiento del síndrome de intestino irritable con estreñimiento (SII-E) en mujeres ≥ de 18 años. Estreñimiento Inducido por Opioides (EIO): AMITIZA está indicado para el tratamiento del estreñimiento inducido por opioides en adultos con dolor crónico no canceroso. No se ha establecido su eficacia en los pacientes tratados con opioides derivados del difenilheptano (p. ej., metadona).
Farmacocinética y farmacodinamia: Farmacocinética: Lubiprostona, después de su administración por vía oral, tiene una disponibilidad sistémica baja y las concentraciones plasmáticas se encuentran por debajo de un nivel de cuantificación (10 pg/mL). Por lo tanto, no es posible calcular de manera confiable los parámetros farmacocinéticos estándar, como área bajo la curva (AUC, area under the curve), concentración máxima (Cmax) y vida media (t½). Sin embargo, sí se han definido los parámetros farmacocinéticos de M3 (el único metabolito activo medible de Lubiprostona). El género no tiene efecto sobre la farmacocinética de M3, después de la administración oral de Lubiprostona.Absorción: Las concentraciones de Lubiprostona en el plasma se encuentran por debajo del nivel de cuantificación (10 pg/mL), lo que se debe a que Lubiprostona tiene una baja disponibilidad sistémica después de su administración oral. Las concentraciones plasmáticas máximas de M3, después de una dosis única de Lubiprostona de 24 mcg, se observan en alrededor de 1.10 horas. La Cmax fue de 41.5 pg/mL y el AUC0-t promedio fue de 57.1 pg• hr/mL. El AUC0-t de M3 aumenta de manera proporcional a la dosis después de dosis únicas de 24 mcg y 144 mcg de Lubiprostona. Distribución: Los estudios in vitro de fijación a proteínas indicaron que cerca del 94% de Lubiprostona se fija a las proteínas plasmáticas humanas. Los estudios en ratas a las que se les administró Lubiprostona radiomarcada indicaron una distribución mínima más allá de los tejidos gastrointestinales. Las concentraciones de Lubiprostona radiomarcada a las 48 horas después de la administración fueron mínimas en todos los tejidos de las ratas. Metabolismo: Los resultados tanto en humanos como en animales indican que Lubiprostona se metaboliza rápida y ampliamente, mediante la reducción en la posición 15, beta-oxidación de la cadena a y omega-oxidación en la cadena. Estas biotransformaciones no están mediadas por el sistema del citocromo P450 hepático sino más bien parecen estar mediadas por la ampliamente diseminada carbonil reductasa. El M3, un metabolito de Lubiprostona que se encuentra tanto en humanos como en animales, se forma por la reducción del grupo carbonilo en la mitad 15-hidroxi que consiste en epímeros tanto a-hidroxi como b-hidroxi. M3 constituye menos de 10% de la dosis de Lubiprostona radiomarcada. En los estudios en animales se ha mostrado que el metabolismo de Lubiprostona pasa con rapidez en el estómago y yeyuno, muy probablemente en ausencia de cualquier absorción sistémica. Eliminación: No fue posible detectar Lubiprostona en el plasma; sin embargo, M3 tiene una t½ que va desde 0.9 hasta 1.4 horas. Después de una dosis oral única de 72 mcg de Lubiprostona marcada con 3H, se recuperó en la orina el 60% de la radioactividad total administrada dentro de las primeras 24 horas y el 30% del total de radioactividad administrada se recuperó en las heces a las 168 horas. Lubiprostona y M3 se detectan sólo en cantidades mínimas en las heces de humanos. Efecto del alimento: Se realizó un estudio con una dosis única de 72 mcg de Lubiprostona marcada con 3H, para evaluar el posible efecto de un alimento sobre la absorción, metabolismo y excreción de Lubiprostona. Los parámetros farmacocinéticos de la radioactividad total demostraron que la Cmax disminuyó 55% mientras que el AUC0-∞ no cambió, cuando Lubiprostona se administró con un alimento rico en grasa. La relevancia clínica del efecto de los alimentos en la farmacocinética de lubiprostona no es clara. Sin embargo, lubiprostona se administró con alimentos y agua en la mayoría de los ensayos clínicos. Poblaciones especiales: Insuficiencia renal: Dieciséis sujetos, de 34 a 47 años de edad (8 sujetos con insuficiencia renal severa [aclaramiento de creatinina (CrCl) < 20 ml/min] que requirieron hemodiálisis y 8 sujetos de control con función renal normal [CrCl > 80 ml/min]), recibieron una dosis única oral de 24 mcg de AMITIZA. Después de la administración, las concentraciones plasmáticas de lubiprostona estaban por debajo del límite de cuantificación (10 pg/ml). Las concentraciones plasmáticas de M3 estuvieron dentro del rango de exposición de la experiencia clínica previa con AMITIZA. Insuficiencia hepática: Veinticinco sujetos, de 38 a 78 años de edad (9 con insuficiencia hepática severa [Child-Pugh C], 8 con insuficiencia hepática moderada [Child-Pugh B] y 8 con función hepática normal) recibieron 12 mcg o 24 mcg de AMITIZA en ayuno. Después de la administración, las concentraciones plasmáticas de lubiprostona estuvieron por debajo del límite de cuantificación (10 pg/ml) excepto para dos sujetos. En sujetos con insuficiencia hepática moderada y severa, la Cmax y el AUC0-t del metabolismo activo M3 de la lubiprostona aumentaron, como se muestra en la Tabla 1.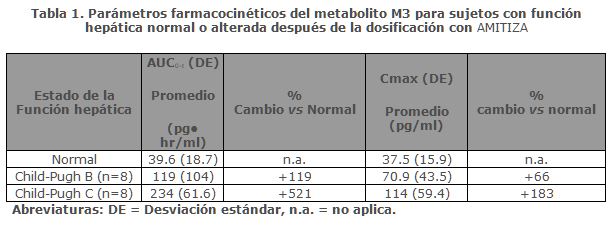
Estos resultados demuestran que existe una correlación entre una mayor exposición de M3 y la severidad de la insuficiencia hepática.Farmacodinamia: Aunque no se han evaluado por completo los efectos farmacológicos de Lubiprostona en humanos, los estudios en animales han mostrado que la administración oral de Lubiprostona aumenta el transporte del ion cloruro hacia la luz intestinal, aumenta la secreción de líquido hacia el intestino y mejora el tránsito fecal.Mecanismo de acción: Lubiprostona un miembro de la clase de compuestos llamados prostonas es un activador de acción local de los canales de cloruro, que mejora la secreción de fluidos intestinales ricos en cloruro, sin alterar las concentraciones séricas de sodio y potasio. Lubiprostona actúa mediante la activación específica de los canales CIC-2, los cuales son un componente normal de la membrana apical del intestino humano, de una manera independiente a la proteína cinasa A.Al aumentar la secreción de líquido intestinal, lubiprostona aumenta la motilidad en el intestino, lo que facilita el paso de las heces y alivia los síntomas asociados con el estreñimiento crónico idiopático. En la técnica de fijación de membrana celular (Patch clamp) en líneas celulares humanas se ha indicado que, la mayor parte de la actividad biológica benéfica de Lubiprostona y sus metabolitos, se observa sólo en la porción apical (luminal) del epitelio gastrointestinal. Lubiprostona, a través de la activación de los canales CIC-2 apicales en las células epiteliales del intestino, elude la acción antisecretora de los opiáceos provocada por la supresión de la excitabilidad de la neurona secretomotora. En estudios ex vivo de intestino isquémico de porcino, se ha mostrado también que la activación del CIC-2 mediante Lubiprostona estimula la recuperación de la función de barrera de la mucosa y reduce la permeabilidad intestinal, a través del restablecimiento de los complejos proteicos de la unión estrecha intercelular. Eficacia clínica: Estreñimiento crónico idiopático: Se realizaron dos estudios doble ciego controlados con placebo, de diseño idéntico en pacientes con estreñimiento crónico idiopático. El estreñimiento crónico idiopático se definió como menos de 3 evacuaciones intestinales espontáneas (SBM) en promedio por semana (una SBM es una evacuación intestinal espontánea, es decir, que ocurre en ausencia del uso de laxantes) junto con uno o más de los siguientes síntomas de estreñimiento por lo menos 6 meses antes de la aleatorización: 1) heces muy duras en al menos una cuarta parte de todas las deposiciones; 2) sensación de evacuación incompleta después de evacuar, en al menos una cuarta parte de todas las deposiciones; y 3) esfuerzo al evacuar al menos en una cuarta parte del tiempo.Después del periodo inicial/lavado de 2 semanas, un total de 479 pacientes (edad promedio 47.2 [rango 20-81] años, 88.9 mujeres, 80.8% caucásicos, 9.6% afroamericanos, 7.3% hispanos, 1.5% asiáticos, 10.9% ≥ 65 años de edad) fueron aleatorizados y recibieron AMITIZA 24 mcg dos veces al día o placebo dos veces al día durante 4 semanas. El objetivo primario de los estudios fue la frecuencia de SBM. Los estudios demostraron que los pacientes tratados con AMITIZA tuvieron una frecuencia más alta de SBM durante la semana 1 que los pacientes con placebo. En ambos estudios, también se observaron resultados similares a los de la Semana 1 en las Semanas 2, 3 y 4 de la terapia (Tabla 2).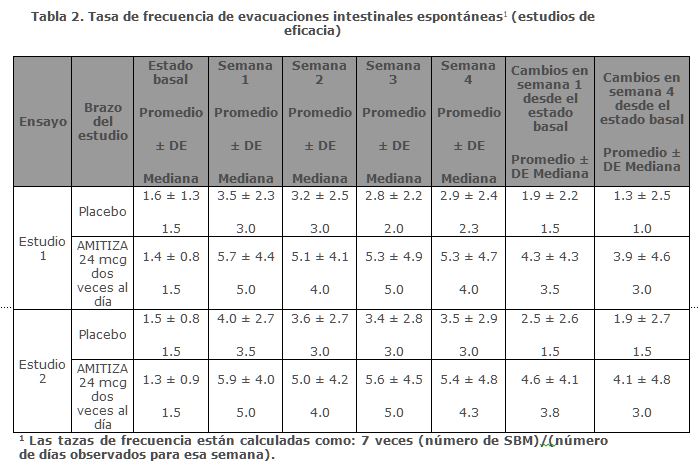
En ambos estudios, AMITIZA mostró incremento en el porcentaje de pacientes que presentaron SBM dentro de las primeras 24 horas después de su administración en comparación con placebo (56.7% vs. 36.9% en el Estudio 1 y 62.9% vs. 31.9% en el Estudio 2, respectivamente). De manera similar, el tiempo hasta la primera SBM fue más corto para los pacientes que recibieron AMITIZA que para los que recibieron placebo. Los signos y síntomas relacionados con el estreñimiento, que incluyen meteorismo, molestia abdominal, cambios en la consistencia de las heces y esfuerzo al evacuar, así como los índices de severidad del estreñimiento, también mejoraron con AMITIZA versus placebo. Los resultados fueron consistentes en los análisis de subpoblación para género, raza y pacientes de la tercera edad (≥ 65 años de edad). Durante un estudio aleatorizado de retiro de 7 semanas, los pacientes que recibieron AMITIZA durante un periodo de tratamiento de 4 semanas fueron aleatorizados para recibir placebo o continuar el tratamiento con AMITIZA. En los pacientes tratados con AMITIZA asignados al azar al placebo, las tasas de frecuencia de SBM volvieron al valor inicial dentro de 1 semana y no provocaron un empeoramiento en comparación con el valor inicial. Los pacientes que continuaron con AMITIZA mantuvieron su respuesta al tratamiento durante las 3 semanas adicionales de tratamiento. Estreñimiento Inducido por Opioides: La eficacia de AMITIZA en el tratamiento del estreñimiento inducido por opioides en pacientes que reciben terapia con opioides para dolor crónico no relacionado con cáncer, se evaluó en tres estudios aleatorizados, doble ciego, controlados con placebo. En el estudio 1, la edad promedio fue de 52 años (rango 20-82) y el 63.1% fueron mujeres. En el estudio 2, la edad promedio fue de 50 años (rango 21-77) y el 64.4% fueron mujeres. En el Estudio 3, la edad promedio fue de 50 años (rango 21-89) y el 60.1% fueron mujeres. Los pacientes habían estado recibiendo terapia opioide estable durante al menos 30 días antes del estudio, que continuaría durante el periodo de tratamiento de 12 semanas. Al inicio del estudio, las dosis orales promedio diarias equivalentes de morfina (MEDD) fueron de 99 mg y 130 mg para los pacientes tratados con placebo y tratados con AMITIZA, respectivamente, en el Estudio 1. Las MEDD promedio al inicio del estudio fueron de 237 mg y 265 mg para los pacientes tratados con AMITIZA y placebo respectivamente, en el Estudio 2. En el Estudio 3, las MEDD promedio al inicio del estudio fueron de 330 mg y 373 mg para los pacientes tratados con placebo y tratados con AMITIZA, respectivamente. El cuestionario Brief Pain Inventoy-Short Form (BPI-SF) se aplicó a los pacientes al inicio del estudio y posteriormente de manera mensual, durante el periodo de tratamiento para evaluar el control del dolor. Los pacientes que habían reportado estreñimiento inducido por opioides al inicio del estudio, definido por tener menos de 3 evacuaciones intestinales espontáneas (SBM) por semana, con al menos 25% de SBM asociadas a una o más de las siguientes condiciones: (1) consistencia de las heces: duras a muy duras; (2) esfuerzo al evacuar de moderado a muy severo; y/o (3) tener sensación de evacuación incompleta. El uso de laxantes se suspendió al comienzo del periodo de detección y durante todo el estudio. Con excepción del periodo de 48 horas antes de la primera dosis y durante al menos 72 horas (Estudio 1) o semana 1 (Estudio 2 y Estudio 3) después de la primera dosis, se permitió el uso de medicamentos de rescate en los casos en los que no se presentaron SBM en un periodo de 3 días. La mediana de las frecuencias semanales de SBM al inicio del estudio fue de 1.5 para pacientes con placebo y 1.0 para los pacientes de AMITIZA en el Estudio 1 y, tanto para el Estudio 2 como para el Estudio 3, la mediana de las frecuencias semanales de SBM al inicio del estudio fueron 1.5 para ambos grupos de tratamiento. En el estudio 1, los pacientes que recibieron opioides distintos al difenilheptano (p. ej., sin metadona) (n = 431) se asignaron al azar para recibir placebo (n = 217) o AMITIZA de 24 mcg dos veces al día (n = 214) durante 12 semanas. El análisis de eficacia primario fue una comparación de la proporción de "pacientes con respuesta general" en cada brazo de tratamiento. Se consideró que un paciente tenía "respuesta general" si presentaba mejoría de ≥1 en la SBM respecto al inicio del estudio y durante todas las semanas de tratamiento para las cuales había datos disponibles y se informaban ≥3 SBM/semana durante al menos 9 de las 12 semanas de tratamiento. La proporción de pacientes en el Estudio 1 que calificó con "respuesta general" fue 27.1% en el grupo que recibió AMITIZA de 24 mcg dos veces al día en comparación con 18.9% de los pacientes que recibieron placebo dos veces al día (diferencia de tratamiento = 8.2%; p=0.03). El análisis de los subgrupos de género y raza no identificó diferencias en la respuesta a AMITIZA entre estos subgrupos. Hubo muy pocos pacientes de la tercera edad (≥ 65 años de edad) para evaluar adecuadamente las diferencias en los efectos en esa población.En el estudio 2, los pacientes en tratamiento con opioides (N = 418) fueron aleatorizados para recibir placebo (n = 208) o AMITIZA de 24 mcg dos veces al día (n = 210) durante 12 semanas. El estudio 2 no excluyó a los pacientes que recibieron opioides derivados del difenilheptano (p. ej., metadona). El objetivo primario de eficacia fue el cambio promedio en la frecuencia de SBM desde el inicio hasta la semana 8; 3.3 vs. 2.4 para AMITIZA y pacientes tratados con placebo, respectivamente; diferencia de tratamiento = 0.9; p = 0.004. La proporción de pacientes en el estudio 2 que califican "con respuesta general", tal como se especificó previamente en el Estudio 1, fue del 24.3% para el grupo que recibió AMITIZA en comparación con el 15.4% para los pacientes que recibieron placebo. En el subgrupo de pacientes del Estudio 2 que tomaron opioides derivados del difenilheptano (Dosis Diaria equivalente a Morfina (MEDD) basal promedio [mediana] fue de 691 [403] mg y 672 [450] mg para pacientes con placebo y AMITIZA, respectivamente), la proporción de pacientes que califican "con respuesta general" fue del 20.5% (8/39) en el grupo que recibió AMITIZA en comparación con el 6.3% (2/32) de los pacientes que recibieron placebo. El análisis de los subgrupos en cuanto a género y raza no identificó diferencias en la respuesta a AMITIZA entre estos subgrupos. Hubo muy pocos pacientes de la tercera edad (≥ 65 años de edad) para evaluar adecuadamente las diferencias en los efectos en esa población.En el Estudio 3, los pacientes que estaban en tratamiento con opioides (N = 451) se asignaron al azar a placebo (n = 216) o a AMITIZA de 24 mcg dos veces al día (n = 235) durante 12 semanas. El estudio 3 no excluyó a los pacientes que recibían opioides derivados del difenilheptano (p. ej., metadona). El objetivo primario de eficacia fue el cambio en la frecuencia de SBM desde el inicio del estudio y hasta la semana 8. El estudio no mostró una mejoría estadísticamente significativa en las frecuencias de SBM en la semana 8 (cambio promedio desde el valor basal de 2.7 comparado con 2.5 para AMITIZA y pacientes tratados con placebo, respectivamente; diferencia en el tratamiento = 0.2, p = 0.76). La proporción de pacientes en el Estudio 3 que califican "con respuesta general", tal como se especificó previamente en el Estudio 1, fue del 15.3% en los pacientes que recibieron AMITIZA en comparación con el 13.0% de los pacientes que recibieron placebo. En el subgrupo de pacientes del Estudio 3 que tomaron opioides derivados del difenilheptano (Dosis Diaria equivalente de Morfina (MEDD) basal promedio [mediana] inicial de 730 [518] mg y 992 [480] mg para los pacientes con placebo y AMITIZA, respectivamente), la proporción de pacientes que califican "con respuesta general" fue del 2.1% (1/47) en el grupo que recibió AMITIZA en comparación con el 12.2% (5/41) de los pacientes que recibieron placebo.Síndrome de Intestino Irritable con Estreñimiento: Se llevaron a cabo dos estudios doble ciego controlados con placebo de diseño similar en pacientes con SII-E. SII se definió como dolor abdominal o malestar que ocurre durante al menos 6 meses con dos o más de los siguientes parámetros: 1) se alivia con la defecación; 2) inicio asociado con un cambio en la frecuencia de las evacuaciones; y 3) inicio asociado con un cambio en la forma de las heces. Los pacientes fueron subclasificados como SII-E si además presentaban dos de las tres siguientes características: 1) < 3 movimientos intestinales espontáneos (SBM) por semana, 2) > 25% heces duras, y 3 > 25% SBM asociadas con esfuerzo al evacuar. Después de 4 semanas del periodo inicial/lavado, un total de 1154 pacientes (edad promedio 46.6 [rango 18-85] años, 91.6% mujeres, 77.4% caucásicos, 13.2% afroamericanos, 8.5% hispanos, 0.4% asiáticos, 8.3% ≥ 65 años de edad) fueron aleatorizados y recibieron AMITIZA de 8 mcg dos veces al día (16 mcg/día) o placebo dos veces al día durante 12 semanas. El objetivo principal de eficacia se evaluó semanalmente mediante la respuesta del paciente a una pregunta general sobre el alivio de síntomas basada en una escala balanceada de 7 puntos ("considerablemente peor" a "considerablemente mejor"): "¿Cómo calificaría el alivio de sus síntomas del SII (molestia/dolor abdominal, hábitos intestinales y otros síntomas del SII) durante la última semana, en comparación a como se sentía antes de ingresar al estudio? El análisis de eficacia primaria fue una comparación de la proporción de quienes presentaron "respuesta general" en cada brazo. Un paciente se consideró con "respuesta general" si los criterios para ser designado con "respuesta mensual" se cumplían en al menos 2 de los 3 meses del estudio. Con "respuesta mensual" se definió a aquel paciente que había informado "alivio considerable" durante al menos 2 semanas del mes o al menos "alivio moderado" en las 4 semanas del mismo mes. Durante cada periodo de evaluación mensual, los pacientes que informaron "empeoramiento moderado" o "empeoramiento notable", aumento en el uso de medicamentos de rescate, o aquellos que suspendieron el tratamiento debido a falta de eficacia, se consideraron sin respuesta.El porcentaje de pacientes que se calificó con "respuesta general" en el estudio 1, fue del 13.8% para el grupo que recibió AMITIZA de 8 mcg dos veces al día en comparación con el 7.8% en pacientes que recibieron placebo dos veces al día. En el estudio 2, el 12.1% de los pacientes del grupo de AMITIZA de 8 mcg presentaron "respuesta general" versus 5.7% de los pacientes del grupo placebo. En ambos estudios, la diferencia entre los tratamientos, placebo y AMITIZA, fue estadísticamente significativa. Resultados en hombres: Los dos estudios aleatorizados, controlados con placebo y doble ciego, incluyeron a 97 pacientes varones (8.4%), lo que es insuficiente para determinar si los hombres con SII-E respondieron de manera diferente a AMITIZA que las mujeres.Durante las 4 semanas posteriores al periodo de retiro del estudio 1, los pacientes que recibieron AMITIZA en el periodo de tratamiento de 12 semanas fueron reasignados al azar para recibir placebo o continuar el tratamiento con AMITIZA. En los pacientes tratados con AMITIZA que presentaron "respuesta total" durante el estudio 1 y que fueron reasignados aleatoriamente para recibir placebo, las tasas de frecuencia de SBM no resultaron en empeoramiento en comparación con el valor inicial.
Contraindicaciones: AMITIZA está contraindicado en pacientes con obstrucción gastrointestinal mecánica conocida o sospechada. AMITIZA está contraindicado en pacientes con alergia conocida a Lubiprostona. Embarazo, lactancia y menores de 18 años.
Precauciones generales: Náuseas: En ocasiones, los pacientes que toman AMITIZA pueden presentar náuseas. La administración concomitante de alimento con AMITIZA puede reducir las náuseas.Diarrea: AMITIZA no debe prescribirse en pacientes que tienen diarrea intensa. Los pacientes deben estar conscientes de la posible aparición de diarrea durante el tratamiento. Debe indicarse a los pacientes que suspendan AMITIZA e informen a su médico si ocurre diarrea grave.Disnea: En los estudios clínicos, se informó de disnea en los pacientes que recibieron AMITIZA. Existen informes postcomercialización de disnea cuando se usa AMITIZA 24 mcg dos veces al día. Algunos pacientes han suspendido el tratamiento debido a la disnea. Estos eventos suelen describirse como una sensación de opresión en el tórax y dificultad para tomar aire y, por lo general, tienen un inicio agudo dentro de los primeros 30 y 60 minutos después de tomar la primera dosis. Por lo general, se resuelve en unas cuantas horas después de tomar la dosis, aunque frecuentemente se ha reportado recurrencia con las dosis posteriores.Obstrucción intestinal: En los pacientes con síntomas que indiquen obstrucción gastrointestinal mecánica, el médico tratante deberá realizar una evaluación minuciosa para confirmar la ausencia de una obstrucción, antes de iniciar la terapia con AMITIZA.Síncope e hipotensión: Se ha informado sobre síncope e hipotensión con AMITIZA en la post-comercialización y algunas de estas reacciones adversas resultaron en hospitalización. La mayoría de los casos ocurrieron en pacientes que tomaban 24 mcg dos veces al día y algunos ocurrieron una hora después de tomar la primera dosis o dosis posteriores de AMITIZA. Algunos pacientes tuvieron diarrea o vómitos concomitantemente antes de desarrollar la reacción adversa. El síncope y la hipotensión generalmente se resuelven después de la interrupción de AMITIZA o antes de la siguiente dosis, pero se ha informado recurrencia con dosis posteriores. Varios casos informaron el uso concomitante de medicamentos conocidos por disminuir la presión arterial lo que puede aumentar el riesgo de desarrollar síncope o hipotensión. Los pacientes deben ser conscientes del riesgo de síncope o hipotensión durante el tratamiento y de que otras reacciones adversas pueden aumentar este riesgo, como diarrea o vómitos.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: Categoría C en el embarazo: Resumen sobre el riesgo: La seguridad de AMITIZA en el embarazo no se ha evaluado en estudios adecuados bien controlados en humanos. AMITIZA deberá utilizarse durante el embarazo sólo si el beneficio potencial justifica el riesgo para el feto.Se observó un aumento en la pérdida fetal, dependiente de la dosis, en conejillos de indias gestantes que recibieron lubiprostona a dosis equivalentes entre 0.2 y 6 veces la dosis máxima recomendada en humanos (DMRH) en función del área de superficie corporal (mg/m2). Los estudios en animales no mostraron un aumento en malformaciones estructurales.Consideraciones clínicas: La experiencia con AMITIZA durante el embarazo es limitada.Actualmente, los datos disponibles sugieren que el aborto espontáneo ocurre de un 15 a un 18% de los embarazos clínicamente reconocidos, independientemente de la exposición a algún fármaco. Considere los riesgos y beneficios de las terapias disponibles cuando trate a una mujer embarazada con estreñimiento crónico idiopático, estreñimiento inducido por opioides o síndrome de intestino irritable con estreñimiento.Estudios de toxicidad:Datos en animales: En estudios de teratogenicidad, ratas y conejos gestantes recibieron lubiprostona oral durante la organogénesis a dosis de hasta 338 veces (ratas) y aproximadamente 34 veces (conejos) la dosis máxima recomendada en humanos (DMRH) en función del área de superficie corporal (mg/m2). Las dosis máximas en animales fueron 2000 mcg/kg/día (ratas) y 100 mcg/kg/día (conejos). En ratas, hubo una mayor incidencia de resorciones tempranas y malformaciones de los tejidos blandos (situs inversus, paladar hendido) con dosis de 2000 mcg/kg/día; sin embargo, estos efectos fueron probablemente secundarios a la toxicidad materna. Un aumento en la pérdida fetal, dependiente de la dosis ocurrió en conejillos de indias que recibieron lubiprostona después del periodo de organogénesis, en los días 40 a 53 de la gestación, a dosis orales diarias de 1, 10 y 25 mcg/kg/día (aproximadamente 0.2, 2 y 6 veces la DMRH en función del área de superficie corporal (mg/m2). El potencial de lubiprostona para causar pérdida fetal también se examinó en monos Rhesus gestantes. Los monos recibieron lubiprostona después de la organogénesis en los días de gestación 110 al 130 a dosis orales diarias de 10 y 30 mcg/kg/día (aproximadamente 3 y 10 veces la DMRH en función del área de superficie corporal (mg/m2). Se observó pérdida fetal en un mono del grupo que recibió dosis de 10 mcg/kg, lo cual está dentro de las tasas históricas normales para esta especie. No se observaron efectos adversos relacionados al fármaco en monos.Lactancia: En ratas, después de la administración oral, no se detectó Lubiprostona ni su metabolito activo en la leche materna. Sin embargo, no se sabe si el fármaco se excreta en la leche materna en humanos. Por lo tanto, debe tenerse cautela cuando se administre AMITIZA a madres en lactancia.Debido a que lubiprostona aumenta la secreción de líquidos en el intestino y la motilidad intestinal, los bebés alimentados con leche materna deben vigilarse para detectar diarrea.
Reacciones secundarias y adversas: Las siguientes reacciones adversas se describen a continuación: Náuseas, Diarrea, Síncope e hipotensión ,Disnea.Datos de los estudios clínicos: Debido a que los estudios clínicos se realizan en condiciones muy variables, las tasas de reacciones adversas observadas en los estudios clínicos de un medicamento no se pueden comparar directamente con las tasas en los estudios clínicos de otro medicamento y pueden no reflejar las tasas observadas en la práctica.Durante el desarrollo clínico de AMITIZA para el ECI, EIO y SII-E, 1234 pacientes fueron tratados con AMITIZA durante 6 meses y 524 pacientes fueron tratados durante 1 año (no se excluyen mutuamente).Estreñimiento Crónico Idiopático: Reacciones adversas en los estudios clínicos realizados para establecer dosis, de eficacia y a largo plazo: Los datos que se describen a continuación reflejan la exposición a AMITIZA de 24 mcg dos veces al día en 1113 pacientes con estreñimiento crónico idiopático durante periodos de tratamiento durante 3 ó 4 semanas, 6 y 12 meses; y datos de 316 pacientes que recibieron placebo durante la exposición a corto plazo (≤ 4 semanas). La población que recibió placebo (N = 316) tenía una edad promedio de 47.8 (rango 21-81) años; 87.3% mujeres; 80.7% caucásicos, 10.1% afroamericanos, 7.3 hispanos, 0.9% asiáticos; y 11.7% de la tercera edad (≥ 65 años de edad). De los pacientes tratados con AMITIZA 24 mcg dos veces al día (N = 1113), la edad promedio fue de 50.3 (rango 19-86) años; 86.9% eran mujeres; 86.1% caucásicos, 7.6% afroamericanos, 4.7% hispanos, 1.0% asiáticos; y 16.7% de la tercera edad (≥ 65 años de edad). La tabla 3 presenta los datos de las reacciones adversas que ocurrieron en al menos 1% de los pacientes que recibieron AMITIZA de 24 mcg dos veces al día y que ocurrieron con mayor frecuencia con el fármaco del estudio comparado con placebo.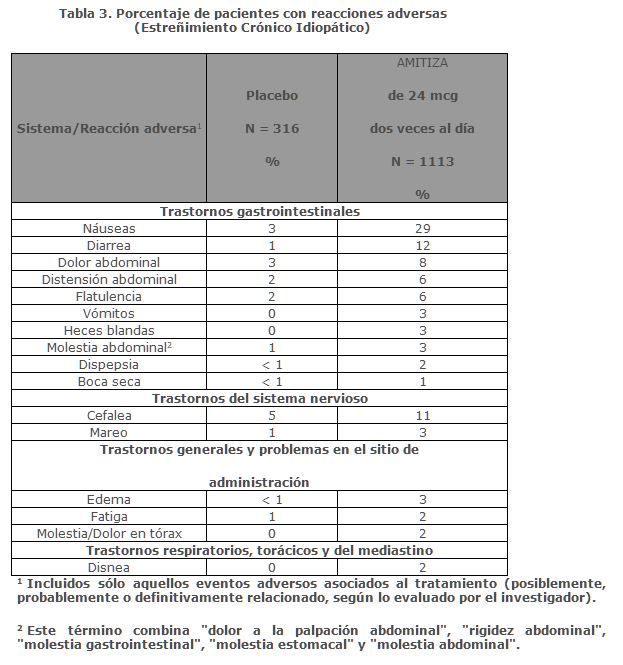
Las reacciones adversas más frecuentes (incidencia > 4%) en ECI fueron náuseas, diarrea, cefalea, dolor abdominal, distensión abdominal y flatulencia.Náuseas: Aproximadamente el 29% de los pacientes que recibieron AMITIZA de 24 mcg dos veces al día presentaron náuseas; el 4% de los pacientes tenía náuseas graves y el 9% de los pacientes suspendió el tratamiento debido a náuseas. La tasa de náuseas asociadas con AMITIZA de 24 mcg dos veces al día fue menor entre los pacientes varones (8%) y de la tercera edad (19%). Ningún paciente en los estudios clínicos fue hospitalizado debido a náuseas.Diarrea: Aproximadamente el 12% de los pacientes que recibieron AMITIZA 24 mcg dos veces al día presentaron diarrea. El 2% de los pacientes tuvo diarrea severa y el 2% de los pacientes suspendieron el tratamiento debido a la diarrea.Electrólitos: No se informaron reacciones adversas graves de desequilibrio electrolítico en estudios clínicos, y no se observaron cambios clínicamente significativos en los niveles de electrólitos séricos en pacientes que recibieron AMITIZA.Reacciones adversas poco frecuentes: Las siguientes reacciones adversas (evaluadas por el investigador como probablemente o definitivamente relacionadas con el tratamiento) ocurrieron en menos del 1% de los pacientes que recibieron AMITIZA de 24 mcg dos veces al día en estudios clínicos en al menos dos pacientes y con mayor frecuencia en pacientes que recibieron el fármaco del estudio en comparación con los que recibieron placebo: incontinencia anal, espasmos musculares, necesidad apremiante de defecar, movimientos intestinales frecuentes, hiperhidrosis, dolor-laríngeo, trastornos gastrointestinales funcionales, ansiedad, sudor frío, estreñimiento, tos disgeusia, eructos, influenza, hinchazón articular, mialgia, dolor, síncope, temblor, apetito disminuido.Estreñimiento Inducido por Opioides: Reacciones adversas en estudios clínicos de eficacia y a largo plazo: Los datos descritos a continuación reflejan la exposición a AMITIZA de 24 mcg dos veces al día en 860 pacientes con EIO durante 12 meses y de 632 pacientes que recibieron placebo dos veces al día durante 12 semanas. La población total (N = 1492) tenía una edad promedio de 50.4 (rango 20-89) años; 62.7% eran mujeres; 82.7% caucásicos, 14.2% afroamericanos, 0.8% indios americanos/nativos de Alaska, 0.8% asiáticos; 5.2% eran de origen étnico hispano; y 8.8% eran de la tercera edad (≥ 65 años de edad). La Tabla 4 presenta los datos de las reacciones adversas que ocurrieron en al menos 1% de los pacientes que recibieron AMITIZA 24 mcg dos veces al día y que ocurrieron con mayor frecuencia con el medicamento del estudio comparado con placebo.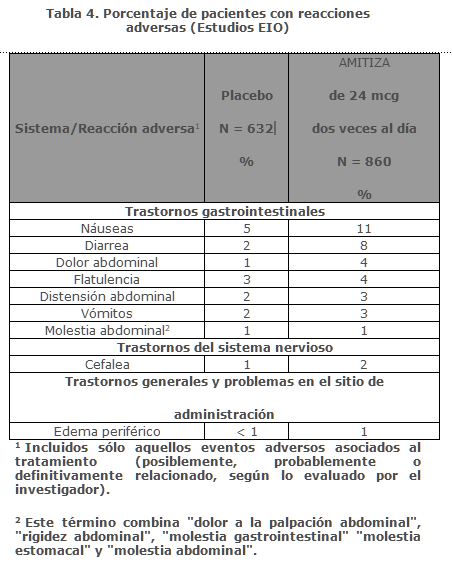
Las reacciones adversas más frecuentes (incidencia > 4%) el EIO fueron náuseas y diarrea.Náuseas: Aproximadamente el 11% de los pacientes que recibieron AMITIZA de 24 mcg dos veces al día presentaron náuseas; el 1% de los pacientes presentó náuseas severas y el 2% de los pacientes interrumpió el tratamiento debido a náuseas.Diarrea: Aproximadamente el 8% de los pacientes que recibieron AMITIZA 24 mcg dos veces al día presentaron diarrea; el 2% de los pacientes tuvo diarrea severa y el 1% de los pacientes suspendieron el tratamiento debido a diarrea.Reacciones adversas poco frecuentes: Las siguientes reacciones adversas (evaluadas por el investigador como probablemente o definitivamente relacionadas con el tratamiento) ocurrieron en menos del 1% de los pacientes que recibieron AMITIZA de 24 mcg dos veces al día en estudios clínicos, en al menos dos pacientes y con mayor frecuencia en pacientes que recibieron el fármaco del estudio en comparación con los que recibieron placebo: incontinencia anal, potasio disminuido en sangre.Síndrome del Intestino Irritable con estreñimiento:Reacciones adversas en los estudios clínicos realizados para establecer dosis de eficacia y a largo plazo: Los datos descritos a continuación reflejan la exposición a AMITIZA de 8 mcg dos veces al día en 1011 pacientes con SII-E durante 12 meses y 435 pacientes que recibieron placebo dos veces al día durante 16 semanas. La población total (N= 1267) tenía una edad promedio de 46.5 (rango 18-85) años; 91.6% eran mujeres; 77.5% caucásicos, 12.9% afroamericanos, 8.6% hispanos, 0.4% asiáticos; y 8.0% de la tercera edad (≥ 65 años de edad).La tabla 5 presenta los datos de las reacciones adversas que ocurrieron en al menos 1% de los pacientes que recibieron AMITIZA 8 mcg dos veces al día y que ocurrieron con mayor frecuencia con el fármaco del estudio que con placebo.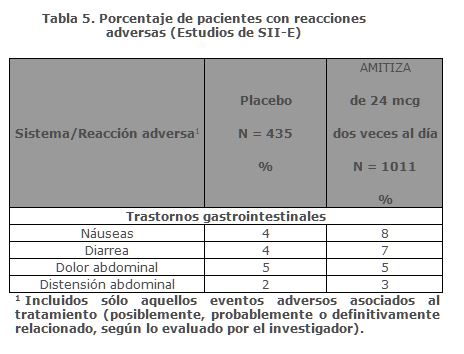
Las reacciones adversas más frecuentes (incidencia > 4%) en SII-E fueron náuseas, diarrea y dolor abdominal.Náuseas: Aproximadamente el 8% de los pacientes que recibieron AMITIZA de 8 mcg dos veces al día experimentaron náuseas; el 1% de los pacientes tuvo náuseas severas y el 1% de los pacientes interrumpió el tratamiento debido a náuseas.Diarrea: Aproximadamente el 7% de los pacientes que recibieron AMITIZA de 8 mcg dos veces al día experimentaron diarrea; < 1% de los pacientes tuvieron diarrea severa y < 1% de los pacientes suspendieron el tratamiento debido a diarrea.Reacciones adversas menos frecuentes: Las siguientes reacciones adversas (evaluadas por el investigador como probablemente relacionadas con el tratamiento) ocurrieron en menos del 1% de los pacientes que recibieron AMITIZA de 8 mcg dos veces al día en estudios clínicos, en al menos dos pacientes y con mayor frecuencia en pacientes que estaban recibiendo el fármaco del estudio que aquellos que recibieron placebo: dispepsia, heces blandas, vómitos, fatiga, boca seca, edema, alanina aminotransferasa elevada, aspartato aminotransferasa elevada, estreñimiento, eructos, enfermedad por reflujo gastroesofágico, disnea, eritema, gastritis, peso aumentado, palpitaciones, infección del tracto urinario, anorexia, ansiedad, depresión, incontinencia anal, fibromialgia, heces duras, letargia, hemorragia rectal, polaquiuria.Experiencia post-comercialización: Se han identificado las siguientes reacciones adversas adicionales durante el uso posterior a la aprobación de AMITIZA. Debido a que estas reacciones se informan voluntariamente a partir de una población de tamaño incierto, no siempre es posible estimar con fiabilidad su frecuencia o establecer una relación causal con la exposición al fármaco.Los informes voluntarios de reacciones adversas que ocurren con el uso de AMITIZA incluyen los siguientes: síncope y/o hipotensión, colitis isquémica, reacciones de hipersensibilidad/reacciones de tipo alérgico (incluyendo erupción, hinchazón y sensación de opresión en la garganta), malestar general, taquicardia, espasmos musculares o espasmos musculares y astenia.
Interacciones medicamentosas y de otro género: No se han realizado estudios de interacción fármaco-fármaco in vivo con AMITIZA.Con base en los resultados de los estudios con microsomas humanos in vitro, existe una baja probabilidad de interacciones farmacocinéticas fármaco-fármaco. Los estudios in vitro que usan microsomas hepáticos humanos indican que las isoenzimas del citocromo P450 no están involucradas en el metabolismo de lubiprostona. Otros estudios in vitro indican que la carbonil reductasa microsómica puede estar implicada en la amplia biotransformación de lubiprostona al metabolito M3. Además, los estudios in vitro en microsomas hepáticos humanos demuestran que lubiprostona no inhibe las isoformas 3A4, 2D6, 1A2, 2A6, 2B6, 2C9, 2C19 o 2E1 del citocromo P450, y los estudios in vitro de cultivos primarios de hepatocitos humanos no muestran inducción del citocromo P450 en las isoformas P450 1A2, 2B6, 2C9 y 3A4 por lubiprostona. Con base en la información disponible, no se prevén interacciones medicamentosas mediadas por la unión a proteínas de importancia clínica.Potencial de interacción con opioides derivados del difenilheptano (por ejemplo, metadona): estudios no clínicos han demostrado que los opioides de la clase química de difenilheptano (por ejemplo, metadona) reducen de forma dosis dependiente la activación de CIC-2 por lubiprostona en el tracto gastrointestinal. Existe la posibilidad de una disminución dependiente de la dosis en la eficacia de AMITIZA en pacientes que usan opioides derivados del difenilheptano.
Alteraciones en los resultados de pruebas de laboratorio: No se dispone de información.
Precauciones y relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Carcinogénesis: Se realizaron dos estudios de carcinogénesis, de dos años de duración, por administración oral forzada (uno en ratones Crl: B6C3F1 y uno en ratas Sprague-Dawley) con Lubiprostona, en dosis de 25, 75, 200 y 500 mcg/kg/día (aproximadamente 2, 6, 17 y 42 veces la dosis más alta recomendada en humanos, respectivamente, con base en el área de superficie corporal). En el estudio de carcinogénesis en ratones, no existió un aumento significativo en la incidencia de tumores. En el estudio de carcinogénesis de dos años en ratas, Lubiprostona en dosis de 20, 100 y 400 mcg/kg/día (aproximadamente 3, 17 y 68 veces la dosis más alta recomendada en humanos, respectivamente, con base en el área de superficie corporal). Existió un aumento significativo de adenoma benigno de células intersticiales de los testículos en las ratas macho con la dosis de 400 mcg/kg/día. En las ratas hembra, el tratamiento con Lubiprostona produjo adenoma hepatocelular con la dosis de 400 mcg/kg/día.Mutagénesis: Lubiprostona no fue genotóxico en el ensayo in vitro de mutación inversa de Ames, el ensayo in vitro de mutación primaria de linfoma de ratón (L5178Y TK+/-), en ensayo in vitro de aberración cromosómica de pulmón de hámster chino (CHL/IU) y en ensayo in vivo de micronúcleo de médula ósea de ratón.Toxicidad reproductiva y de desarrollo: Lubiprostona, en dosis orales de hasta 1,000 mcg/kg/día, no tuvieron efecto en la fertilidad y función reproductiva de ratas macho y hembra. Sin embargo, el número de sitios de implante y los embriones vivos se redujeron significativamente en las ratas en el grupo de dosis de 1,000 mcg/kg/día, en comparación con el grupo control. El número de embriones muertos o reabsorbidos en el grupo de 1,000 mcg/kg/día fue mayor en comparación con el grupo control, pero no fue estadísticamente significativo. La dosis de 1,000 mcg/kg/día en las ratas es aproximadamente 169 veces la dosis más alta recomendada en humanos de 48 mcg/día con base en el área de superficie corporal.En conejos qu