APROVEL®
SANOFI AVENTIS
Denominación genérica: Irbesartan.
Forma farmacéutica y formulación: Tabletas. Cada tableta contiene: irbesartan 75 mg, 150 mg, 300 mg. Excipiente cbp 1 tableta.
Indicaciones terapéuticas: APROVEL® está indicado para el tratamiento de la hipertensión arterial. Se puede utilizar ya sea solo o en combinación con otros agentes antihipertensivos tales como: diuréticos del tipo de las tiazidas, agentes beta-bloqueadores y agentes bloqueadores de los canales del calcio de acción prolongada. APROVEL® también está indicado para la reducción de la progresión de la nefropatía y protección renal en pacientes con diabetes mellitus tipo II.
Farmacocinética y farmacodinamia: APROVEL® (irbesartan) es un antagonista no péptido de los receptores de la angiotensina II (subtipo AT1). Está disponible en forma de tabletas de 75, 150 y 300 mg para administración oral. Mecanismo de acción: irbesartan es un antagonista específico de los receptores de la angiotensina II (subtipo AT1). La angiotensina II es un componente importante del sistema renina-angiotensina y participa en la fisiopatología de la hipertensión y en la homeostasis del sodio. Irbesartan no requiere de activación metabólica para su acción. Bloquea los potentes efectos de vasoconstricción y de secreción de aldosterona y de angiotensina II, por antagonismo selectivo de los receptores de la angiotensina II (subtipo AT1) localizados en las células del músculo liso vascular y en la corteza suprarrenal. No tiene actividad agonista en el receptor AT1 y tiene una afinidad mucho mayor (más de 8.500 veces) para el receptor AT1 que para el receptor AT2 (receptor que ha demostrado asociación con la homeostasis cardiovascular). Irbesartan no inhibe a las enzimas que participan en el sistema renina-angiotensina es decir, la enzima convertidora de angiotensina (ECA) ni afecta a otros receptores de hormonas o canales de iones que intervienen en la regulación cardiovascular de la tensión arterial y en la homeostasis del sodio. El bloqueo de los receptores AT1 causado por irbesartan interrumpe el circuito de retroalimentación dentro del sistema renina-angiotensina-aldosterona, lo que resulta en incrementos de los niveles plasmáticos de renina y angiotensina II. Después de la administración de irbesartan declinan las concentraciones de aldosterona en plasma, sin embargo, los niveles de potasio en suero no se afectan de manera significativa (incremento medio de < 0,1 mEq/l) con las dosis recomendadas. Irbesartan no tiene efectos notables sobre las concentraciones séricas de triglicéridos, colesterol o glucosa; no tiene tampoco efectos sobre el ácido úrico del suero ni sobre la excreción urinaria del ácido úrico. Propiedades farmacocinéticas: irbesartan es un agente activo que se administra por vía oral y que no requiere biotransformación para su acción. Luego de la administración oral, irbesartan se absorbe rápida y completamente. La biodisponiblidad absoluta de irbesartan administrado por la vía oral es de 60-80%. Los alimentos no afectan la biodisponibilidad. Las concentraciones pico en plasma se presentan de 1.5 a 2 horas después de su administración oral. Irbesartan se une a las proteínas del plasma en un 96% y tiene un enlace no significativo con los componentes celulares de la sangre. El volumen de distribución es de 53-93 litros. En el plasma, el irbesartan sin cambios representa el 80-85% de la radioactividad circulante después, de la administración oral o intravenosa de irbesartan marcado con C14. Irbesartan se metaboliza en el hígado vía conjugación del glucurónido y oxidación. Su principal metabolito circulante es irbesartan glucurónido (~6%). Irbesartan experimenta oxidación, principalmente por la isoenzima CYP2C9 citocromo P450; la isoenzima CYP3A4 tiene un efecto insignificante. Irbesartan no es metabolizado por la mayoría de las isoenzimas que intervienen comúnmente en el metabolismo del fármaco, ni las induce o inhibe de manera substancial (esto es CYP1A1, CYP1A2, CYP2A6, CYP2B6, CYP2D6 o CYP2E1). Irbesartan no induce ni inhibe a las isoenzima CYP3A4. Irbesartan y sus metabolitos se excretan por vía biliar y por vía renal. Alrededor del 20% de la radioactividad administrada después de una dosis de irbesartan C14 por vía oral o intravenosa se recupera en la orina, y el resto en las heces. Menos del 2% de la dosis se excreta en la orina como irbesartan sin cambio. La vida media de eliminación terminal (t ½) de irbesartan es de 11-15 horas. La eliminación total del organismo de irbesartan administrado por vía intravenosa es de 157-76 ml/min, de los cuales 3,0 a 3,5 ml/min son por eliminación renal. Irbesartan exhibe una farmacocinética lineal sobre los límites de la dosis terapéutica. Las concentraciones en estado estable en plasma se alcanzan dentro de los tres días del inicio del régimen de dosificación una vez al día. Se observó acumulación limitada ( < 20%) en plasma al repetir la dosis diaria. En individuos hipertensos se observaron concentraciones más altas de irbesartan en plasma (11-44%) en las mujeres que en los hombres; sin embargo, después de múltiples dosis, no se observaron diferencias en cuanto a acumulación o vida media de eliminación entre hombres y mujeres. No se han observado diferencias específicas de género respecto al efecto clínico. En sujetos (hombres y mujeres) normotensos ancianos (65-80 años) con función renal y hepática clínicamente normal, el AUC y las concentraciones pico en plasma (Cmáx) de irbesartan fueron aproximadamente 20 a 50% mayores que las que se observaron en los sujetos más jóvenes (18 a 40 años). Sin considerar la edad, la vida media de eliminación es semejante. No se han observado diferencias significativas relacionadas con la edad respecto al efecto clínico. En sujetos normotensos negros y blancos, el AUC en plasma y la (t ½) de irbesartan son aproximadamente 20 a 50% mayores en los negros que en los blancos; las concentraciones pico en plasma (Cmáx) de irbesartan fueron básicamente equivalentes. En pacientes con deterioro renal (sin considerar el grado) y en pacientes en hemodiálisis la farmacocinética de irbesartan no se alteró significativamente. Irbesartan no es removido mediante hemodiálisis. En los pacientes con insuficiencia hepática debida a cirrosis leve a moderada no se alteró significativamente la farmacocinética de irbesartan. Propiedades farmacodinámicas: el efecto de disminución de la tensión arterial de irbesartan se hace aparente después de la primera dosis y está presente de manera importante durante 1-2 semanas; el efecto máximo ocurre en 4-6 semanas. En estudios de seguimiento a largo plazo el efecto de irbesartan se mantuvo durante más de un año. Una sola dosis al día hasta de 900 mg produjo descensos de la tensión arterial dosis-relacionados. Las dosis de 150-300 mg una vez al día disminuyen la tensión arterial en posición supina o sedente, en cubeta (esto es 24 horas después de tomar la dosis), en un promedio de 8-13/5-8 mmHg (sistólica/diastólica) cifras más altas que aquellas disminuciones causadas por placebo. Los efectos en cubeta son el 60-70% de las repuestas diastólicas y sistólicas pico correspondientes. Los efectos óptimos sobre la tensión arterial durante las 24 horas se obtienen con la administración de la dosis una vez al día. La tensión arterial disminuye aproximadamente en el mismo grado tanto en la posición de pie como en la posición supina. Los efectos ortostáticos no son frecuentes, pero igual que con los inhibidores de ECA, se puede esperar que ocurran en pacientes que tienen depleción de sodio y/o depleción de volumen. Los efectos de disminución de la tensión arterial de irbesartan y los efectos de los diuréticos del tipo de las tiazidas se suman. En los pacientes que no se controlan adecuadamente con irbesartan solo, la adición de una dosis baja de hidroclorotiazida (12,5 mg) al irbesartan una vez al día resulta en una mayor reducción en cubeta de la tensión arterial en comparación con placebo de 7-10/3-6 mmHg (sistólica/diastólica). La efectividad de irbesartan no es influenciada por la edad ni por el género. Tal como sucede con otros fármacos que afectan el sistema renina-angiotensina, los pacientes negros tienen una respuesta notablemente menor a la monoterapia con irbesartan. Cuando se administró irbesartan concomitantemente con hidroclorotiazida en dosis bajas (12,5 mg diarios), la respuesta antihipertensiva en los pacientes negros se aproximó a la de los pacientes blancos. Después de la descontinuación de irbesartan, la tensión arterial regresa en forma gradual hacia la línea basal. No se ha observado hipertensión de rebote. Farmacología clinica: estudio de los "Efectos de irbesartan en pacientes hipertensos con diabetes mellitus tipo 2 y microalbuminuria" (IRMA 2), estudio multicéntrico, aleatorizado, controlado con placebo doble ciego, en el que se incluyeron 590 pacientes dignósticados, con microalbuminuria (20-200 mg/min, 30-300 mg/día) y función renal normal (creatinina sérica ≤1,5 mg/dl en hombres y ≤1,1 mg/dl en mujeres). El estudio presentó como objetivo primario evaluar los efectos a largo plazo (2 años) del uso de APROVEL® en la evolución clínica de la proteinuria (rango de excreción de albúmina en orina (AER) > 200 mg/min; > 300 mg (día y el incremento de AER de por lo menos el 30% sobre los datos de inicio). Además, después de 1 a 2 años de tratamiento, se determinaron los cambios de AER durante la noche y el aclaramiento de creatina/24 horas. APROVEL® de 300 mg demostró un 70% en la reducción del riesgo relativo en el desarrollo de proteinuria clínica, al compararse con placebo (p=0,0004). APROVEL® 150 mg demostró un 39% en la reducción de riesgo relativo en el desarrollo de proteinuria, al compararse con placebo (p=0,085). La disminución en la velocidad de la progresión del desarrollo clínico de la proteinuria fue evidente desde los tres meses y continuo durante un período de 2 años. La disminución en la depuración de creatinina/24 horas, no presenta diferencias significativas en los tres grupos estudiados, sin embargo, la normoalbuminuria ( < 20 mg/min; < 30 mg/día) fue reportada con mayor frecuencia en el grupo que manejó dosis de APROVEL® de 300 mg (34%), al compararse con el grupo placebo (21%). En el estudio clínico de ibersartan en Neuropatía diabética (IDNT), estudio multicéntrico, aleatorizado controlado, doble ciego, de morbi-mortalidad, se compararon APROVEL®, amlodipino y placebo. En 1715 pacientes hipertensos con diabetes mellitus tipo 2 (proteinuria ≥ 900 mg/día y creatinina sérica 1,0-3,0 mg/dl) examinándose los efectos a largo plazo (2,6 años) de APROVEL® en la progresión de la enfermedad renal y las causas de mortalidad. Agregando a este estudio un segundo objetivo, determinar los efectos de APROVEL® sobre los riegos de aparición de eventos cardiovasculares fatales o no fatales. Los pacientes fueron divididos aleatoriamente en tres grupos para recibir APROVEL® 75 mg, amlodipina 2,5 mg o placebo 1 vez/día. Los pacientes fueron impregnados hasta alcanzar una dosis de mantenimiento de 300 mg de APROVEL® 10 mg de amlodipino o placebo. APROVEL® demostró un 20% de diferencia al compararse con el grupo placebo en la reducción del riesgo de aparición de los siguientes eventos clínicos: incremento de los niveles de creatinina sérica, enfermedad renal en fase terminal, mortalidad por cualquier causa (p=0,024) y un 23% de riesgo relativo al compararse con el grupo de amlodipino (p=0,006). Datos similares se presentaron al comparar los rangos de presión arterial entre los grupos de APROVEL® y amlodipino. No hay una diferencia significativa en el desarrollo de eventos cardiovasculares fatales o no fatales (muerte, infarto agudo al miocardio no fatal, hospitalización por insuficiencia cardíaca, daño neurológico permanente secundario a Eventos Vasculares Cerebrales (EVC) o amputación), entre los tres grupos en tratamiento. Los efectos de irbersartan en eventos renales no fueron similares al evaluar los subgrupos (mujeres, etnias) participantes: su uso parece ser menos favorable en mujeres y negros. El análisis de los subgrupos fue difícil de interpretar, y no es posible diferenciar si estos efectos son diferencias reales o posibilidades.
Contraindicaciones: APROVEL® está contraindicado en los pacientes que tienen hipersensibilidad al irbesartan o a alguno de los componentes de la fórmula. Embarazo y lactancia. Advertencias: hipotensión: en pacientes con depleción de volumen APROVEL® raramente se ha asociado con hipotensión en los pacientes hipertensos que no tienen otro padecimiento concomitante. Es posible que ocurra hipotensión sintomática, tal como sucede con los inhibidores de la ECA, en los pacientes con depleción de sodio/volumen y en aquellos tratados vigorosamente con diuréticos y/o con restricción de sal o en hemodiálisis. La depleción de volumen y/o sodio debe corregirse antes de iniciar el tratamiento con irbesartan o considerar el inicio del tratamiento con una dosis menor. Morbilidad y mortalidad fetal/neonatal: no obstante que no se tiene experiencia con APROVEL® en mujeres embarazadas, se ha reportado que la exposición del producto a los inhibidores de la ECA administrados a mujeres embarazadas, durante el segundo y tercer trimestres, causa lesiones y muerte del feto. Por lo tanto, al igual que cualquier otro fármaco que actúa directamente sobre el sistema renina-angiotensina-aldosterona, APROVEL® no debe administrarse durante el embarazo. Cuando el embarazo se detecta durante el tratamiento, APROVEL® debe suspenderse tan pronto como sea posible.
Precauciones generales: Como consecuencia de la inhibición del sistema renina-angiotensina-aldosterona, se pueden esperar cambios en la función renal de los individuos susceptibles. En los pacientes cuya función renal depende de la actividad del sistema renina-angiotensina-aldosterona (pacientes hipertensos con estenosis de la arteria renal de uno o de ambos riñones o pacientes con insuficiencia cardíaca congestiva grave), el tratamiento con otros fármacos que afecten este sistema se ha asociado con oliguria y/o azotemia progresiva y raramente con insuficiencia renal aguda y/o el fallecimiento. No se puede excluir la posibilidad de que ocurra un efecto similar con el uso de un antagonista de los receptores de angiotensina II. En caso de pacientes hipertensos con diabetes mellitus tipo 2 con proteinuria (≥ 900 mg/día), población con alto riego de desarrollo de estenosis en la arteria renal, ningún paciente tratado con APROVEL® durante el estudio IDNT tuvo incrementos tempranos agudos de los niveles séricos de creatinina atribuibles a modificaciones en la arteria renal. Uso geriátrico: en los pacientes que recibieron irbesartan en los estudios clínicos no se observaron diferencias generales en cuanto a la eficacia y la seguridad en los pacientes de mayor edad (65 años o mayores) ni en lo pacientes más jóvenes. Uso pediátrico: no se ha establecido la seguridad ni la eficacia de los antagonistas de los receptores de angiotensina II en pacientes pediátricos.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: APROVEL® está contraindicado durante el embarazo. Aunque no existe experiencia con APROVEL® en mujeres embarazadas, la exposición in utero a los inhibidores de la ECA administrados a mujeres embarazadas durante el segundo y tercer trimestre ha reportado daño y muerte fetal. Al igual que cualquier medicamento que actúe sobre el sistema renina-angiotensina-aldosterona, APROVEL® no debe ser usado durante el embarazo. Si el embarazo se presenta durante el tratamiento con APROVEL®, éste deberá suspenderse tan pronto como sea posible. Madres que amamantan: APROVEL® está contraindicado en la lactancia. Irbesartan se excreta en la leche de las ratas que amamantan. No se sabe si irbesartan o sus metabolitos se excretan en la leche humana. Se debe tomar la decisión de interrumpir la lactancia o suspender la administración del fármaco, tomando en consideración la importancia del tratamiento de la madre con APROVEL® y el riesgo potencial para el lactante.
Reacciones secundarias y adversas: Eventos adversos: se ha evaluado APROVEL® en cuanto a seguridad en estudios clínicos con 5.000 sujetos aproximadamente, incluidos 1.300 pacientes hipertensos tratados durante 6 meses y más de 400 pacientes tratados durante un año o más. Por lo general, los eventos adversos en los pacientes que recibieron APROVEL® fueron leves y transitorios y no tuvieron relación con la dosis. La incidencia de eventos adversos no estuvo relacionada con la edad, el género o la raza. En estudios clínicos placebo-controlados, que incluyeron 1.965 pacientes tratados con irbesartan (duración usual del tratamiento de 1 a 3 meses), la descontinuación del tratamiento a causa de algún evento adverso clínico o de laboratorio, fue de 3,3 por ciento para los pacientes tratados con irbesartan y de 4,5 por ciento para los pacientes tratados con placebo (p=0,029). En la tabla que sigue se muestran los eventos adversos clínicos probablemente o posiblemente relacionados, o con una relación incierta con el tratamiento, que ocurrieron en por lo menos en el 1% de los pacientes tratados con irbesartan o con placebo, en los ensayos controlados: eventos adversos clínicos* en ensayos e hipertensión placebo-controlados en hipertensión arterial: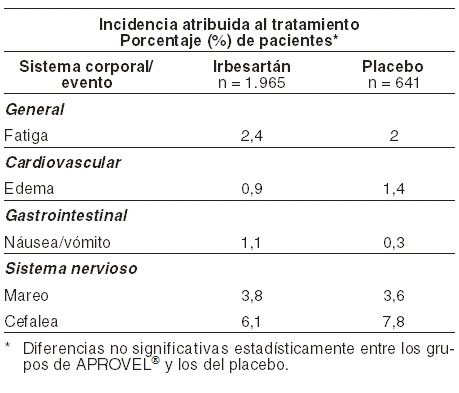
Otros eventos clínicos probablemente o posiblemente relacionados o con una relación incierta con el tratamiento, que ocurrieron con una frecuencia de 0,5% a < 1% y con una incidencia similar o ligeramente mayor en los pacientes tratados con irbesartan que en los pacientes tratados con placebo incluyeron: dolor torácico, tos, diarrea, dispepsia/pirosis, vahídos (ortostáticos), disfunción sexual y taquicardia. Ninguno de los eventos fue significativamente diferente desde el punto de vista estadístico entre los pacientes tratados con irbesartan y los pacientes tratados con placebo. Muy raramente se han reportado casos de hipersensibilidad con irbersartan (angioedema, urticaria). Los siguientes eventos clínicos han sido reportados raramente durante la farmacovigilancia post-comercialización, sin embargo, no se ha establecido una relación causal: astenia, hiperpotasemia, mialgias, ictericia, incremento en los resultados en las pruebas de funcionamiento hepático y alteraciones de la función renal, incluyendo casos aislados de falla renal en pacientes con factores de riesgo. Estudios clínicos en pacientes con hipertensión arterial y enfermedad renal secundaria a diabetes mellitus tipo 2: los eventos clínicos reportados fueron similares a aquellos reportados con estudios clínicos en pacientes hipertensos, con excepción de síntomas ortostáticos (mareos, mareos ortostásticos e hipotensión ortostática), datos obtenidos en el estudio IDNT (proteinuria ≥900 mg/día y creatinina sérica en rangos que varían entre 1,0 a 3,0 mg/dl). Los síntomas ortostáticos ocurrieron más frecuentemente en el grupo de pacientes tratados con APROVEL® (mareo 10,2%, mareo ortostático 5,4%, hipotensión ortostática 5,4%), que en el grupo placebo (mareo 6,0%, mareo ortostático 2,7%, hipotensión ortostática 3,2). Los porcentajes de suspensión del tratamiento secundario a los síntomas ortostáticos del APROVEL® versus placebo fueron: mareos 0,3% vs. 0,5%, mareos ortostáticos 0,2 versus 0,0% hipotensión ortostática 0 versus 0.
Interacciones medicamentosas y de otro género: Con base en la información in vitro, no es de esperarse que ocurran interacciones con fármacos cuyo metabolismo es dependiente de las isoenzimas del citocromo P450 CYP1A1, CYP1A2, CYP2A6, CYP2B6, CYP2E1 o CYP3A4. Irbesartan es metabolizado principalmente por CYP2C9; sin embargo, durante los estudios clínicos de interacciones no se observaron interacciones significativas cuando se administró irbesartan en forma concomitante con warfarina (un medicamento metabolizado por CYP2C9). Irbesartan no afecta la farmacocinética de la digoxina. La farmacocinética de irbesartan no se afecta por la administración concomitante de nifedipina o de hidroclorotiazida. Con base en la experiencia con el uso de otros medicamentos que afectan el sistema renina-angiotensina, la administración de diuréticos ahorradores de potasio, suplementos de potasio o substitutos de sal que contienen potasio puede causar incremento del potasio sérico.
Alteraciones en los resultados de pruebas de laboratorio: En reportes de dos estudios clínicos de pacientes con heprtensión arterial y enfermedad renal secundaria a diabetes mellitus tipo 2 (IDNT e IRMA 2), se encontraron los siguientes resultados: hiperpotasemia: En IDNT, el porcentaje de pacientes con hiperpotasemia ( > 6mEq/l) fue de 18,6% en el grupo APROVEL®, comparado con el 6% del grupo placebo. En el IDNT, el rango de suspensión del tratamiento relacionado con la hiperpotasemia fue de 2,1% en el grupo APROVEL® versus 0,36% del grupo placebo. En el estudio IRMA 2 el porcentaje de sujetos con hiperpotasemia ( > 6mEq/L) fue de 1,0% vs. 0% en el grupo APROVEL® y el rango de suspensión del tratamiento relacionado con hiperpotasemia fue de 0,5% del grupo APROVEL® versus 0% del grupo placebo. Sin embargo, en las determinaciones de diversos parámetros de las pruebas de laboratorio de los estudios clínicos controlados en pacientes con hipertensión, no se presentaron cambios clínicos significativos. No es necesario un monitoreo especial de las parámetros de laboratorio en los pacientes con hipertensión arterial esencial que reciben tratamiento con APROVEL®.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: No se observó evidencia de carcinogenicidad cuando se administró irbesartan en dosis hasta de 500/1.000 mg/kg/día (machos/hembras, respectivamente) a ratas y 1.000 mg/kg/día a ratones durante dos años. Estas dosis produjeron una exposición sistémica 4-25 veces (ratas) y 4-6 veces (ratones) mayor que la exposición en humanos que recibieron 300 mg diarios. Irbesartan no fue mutagénico en una batería de pruebas in vitro (prueba microbiana de Ames, prueba de reparación de DNA en hepatocito de rata, ensayo de mutación de gen prematuro en célula de mamífero V79). Irbesartan fue negativo en varias pruebas para inducción de aberraciones cromosómicas (ensayo de linfocito humano, in vitro; estudio de micronúcleo de ratón, in vitro). La fertilidad y el funcionamiento de la reproducción no fueron afectadas en estudios de ratas machos y hembras, aun con dosis de irbesartan administradas por vía oral que causan alguna toxicidad en los padres (hasta 650 mg/kg/día). No se observaron efectos significativos en la cantidad de cuerpos lúteos, de implantes o de fetos vivos. Irbesartan no afectó la sobrevida, el crecimiento no la reproducción de la descendencia. A dosis de 50 mg/kg/día y mayores se observaron efectos transitorios (incremento de la cavitación de la pelvis renal, hidrouréter o edema subcutáneo) en los fetos de ratas, los cuales se resolvieron después del nacimiento. En conejos se observó mortalidad materna, aborto y resorción fetal temprana, con dosis de 30 mg/kg/día. No se observaron efectos teratogénicos de otro tipo en la rata ni en el conejo.
Dosis y vía de administración: La dosis usual, inicial y de mantenimiento de APROVEL® es de 150 mg una vez al día. Se puede administrar con o sin los alimentos. El tratamiento se debe ajustar de acuerdo con la respuesta de la tensión arterial. A los pacientes que requieren un mayor control de la tensión arterial se les debe incrementar la dosis a 300 mg una vez al día. Cuando la tensión arterial no se controla adecuadamente con APROVEL® solo, se puede agregar un diurético (hidroclorotiazida 12,5 mg diarios) otro agente antihipertensivo (un agente betabloqueador o un agente bloqueador de los canales de calcio de acción prolongada). Pacientes con depleción del volumen intravascular: a los pacientes con depleción grave de volumen y/o con depleción de sodio, tales como aquellos tratados vigorosamente con diuréticos o con hemodiálisis, se les debe corregir este trastorno antes de la administración de APROVEL® o considerarse la administración de una dosis inicial menor. Cuando la tensión arterial no se controla adecuadamente, se puede incrementar la dosis. Ancianos y pacientes con deterioro renal o hepático: por lo general no es necesario reducir la dosificación en los ancianos ni en los pacientes con deterioro de la función renal (independientemente del grado) o con deterioro de la función hepática (de grado leve a moderado). En paciente hipertensos con diabetes tipo II y deterioro renal, APROVEL® 300 mg una vez al día es la dosis sugerida para mantenimiento.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: La experiencia con adultos expuestos a dosis hasta de 900 mg/día por 8 semanas no reveló toxicidad. No se dispone de información específica acerca del tratamiento de la sobredosificación con APROVEL®. Se debe hacer un monitoreo estrecho del paciente y el tratamiento debe ser sintomático y de soporte. Las medidas sugeridas incluyen la inducción del vómito y/o el lavado gástrico. Irbesartan no se remueve del organismo con la hemodiálisis.
Presentación(es): Caja de cartón con 14 y 28 tabletas de 75 mg, 150 mg y 300 mg en envase de burbuja.
Recomendaciones sobre almacenamiento: Consérvese a temperatura ambiente a no más de 30°C y en lugar seco.
Leyendas de protección: Literatura exclusiva para médicos. No se deje al alcance de los niños. Su venta requiere receta médica.
Nombre y domicilio del laboratorio: Sanofi-Aventis de México S.A. de C.V. Oficinas: Av. Universidad No. 1738, 04000, Coyoacán, México, D.F., Planta: Acueducto del Alto Lerma No. 2, Zona Industrial Ocoyoacac, 52740 Ocoyoacac, Edo. de México
Número de registro del medicamento: 415M97 SSA IV.
Clave de IPPA: DEAR-06350122740077/RM 2007