APTIVUS*
BOEHRINGER PM
Cápsulas
Denominación genérica: Tipranavir.
Forma farmacéutica y formulación: Cada cápsula contiene: tipranavir 250 mg. Vehículo cbp 1 cápsula.
Indicaciones terapéuticas: APTIVUS®, coadministrado con ritonavir a dosis bajas, está indicado para el tratamiento antirretroviral de combinación en pacientes adultos infectados con VIH-1 que han tenido experiencia con tratamientos antirretrovirales y para pacientes que están infectados con cepas VIH-1 resistentes a más de un inhibidor de proteasa. Al decidir sobre un nuevo régimen de tratamiento para los pacientes que han fallado al tratamiento con un régimen antirretroviral, debe realizarse una cuidadosa valoración individual del historial de tratamiento del paciente y de los patrones de mutaciones asociados con los diferentes fármacos. Puede ser apropiado utilizar las pruebas de resistencia cuando éstas se encuentren disponibles. Hay datos insuficientes en el tratamiento de pacientes pediátricos y niños con APTIVUS®; por lo tanto, no es recomendado.
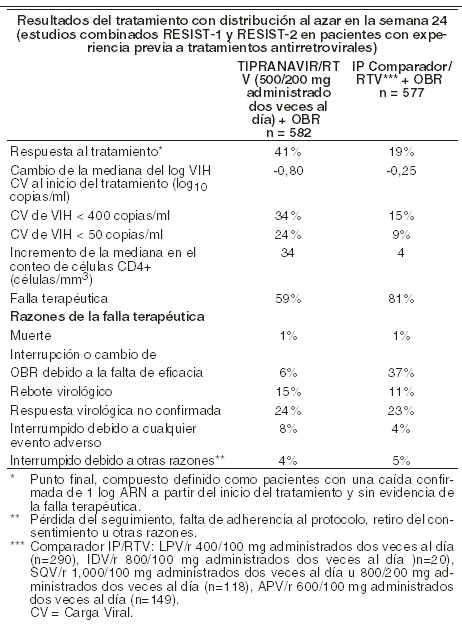
Farmacocinética y farmacodinamia: Mecanismo de acción: el virus de la inmunodeficiencia humana (VIH1) codifica una aspartilo proteasa que es esencial para el corte y la maduración de los precursores de la proteína viral. Tipranavir, principio activo de APTIVUS®, es un inhibidor no peptídico de la proteasa del VIH-1 que inhibe la replicación viral evitando la maduración de las partículas virales. Actividad antiviral in vitro: APTIVUS® inhibe la replicación de las cepas del laboratorio del VIH-1 y los aislados clínicos modelos agudos de infección de células T, con concentraciones efectivas en un 50% (CE50) que varían de 0,03 a 0,07 mm (18-42 ng/ml). APTIVUS® demuestra actividad antiviral in vitro contra un panel extenso de grupo M de VIH-1 no clase B aislados (A, C, D, F, G, H, CRF01 AE, CRF02 AG, CRF12 BF). Grupo O y VIH-2 aislados tienen susceptibilidad reducida in vitro para APTIVUS®, con valores EC50 en rangos desde 0,164 - 1 mm y 0,233 - 0,522 mm, respectivamente. Los estudios de unión a proteínas han demostrado que la actividad antiviral del APTIVUS® disminuye en un promedio de 3,75 veces en condiciones donde se encuentra presente el suero humano. Cuando se utilizó otro agente antirretroviral in vitro, la combinación de APTIVUS® fue como aditivo antagonista con otros inhibidores de proteasa (amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir y saquinavir) y aditivos generalmente con el NNRTI's (delavirdina, efavirenz y nevirapina) y los NRTI's (abacavir, didanosina, emtricitabina, lamivudina, stavudina, tenofovir y zidovudina). El inhibidor de la fusión de HIV enfurviritida fue sinérgico con APTIVUS®. Esto no fue antagonismo de combinaciones in vitro de APTIVUS® con cualquiera de adefovir o ribavirina, usados en el tratamiento de hepatitis viral. Resistencia: el desarrollo de la resistencia in vitro a APTIVUS® es lento y complejo. En un experimento particular de resistencia in vitro, se seleccionó un aislado del VIH-1 que fue 87 veces más resistente al APTIVUS® después de 9 meses, y contenía 10 mutaciones en la proteasa. L10F, I13V, V32I, L33F, M36I, K45I, I54V/T, A71V, V82L, I84V, así como una mutación en el sitio de corte CA/P2 de la poliproteína gag. Experimentos genéticos inversos demostraron que fue necesaria la presencia de 6 mutaciones en la proteasa (I13V, V32I, L33F, K45I, V82L, I84V) para conferir > 10 veces la resistencia al APTIVUS®, mientras que el genotipo de mutación 10 confirió una resistencia de 69 veces a APTIVUS®. Existe una correlación inversa in vitro entre el grado de resistencia a APTIVUS® y la capacidad de los virus para replicarse. Los virus recombinantes que muestran una resistencia ≥3 veces a APTIVUS® crecen menos del 1% de la relación detectada para el VIH-1 tipo salvaje en las mismas condiciones. A través de una serie de múltiples análisis de regresión escalonada de genotipos al inicio del tratamiento y durante éste a partir de todos los estudios clínicos, se han asociado 16 aminoácidos con una reducción a la susceptibilidad al APTIVUS® y/o reducción de la respuesta de la carga viral a las 24 semanas: 10V, 13V, 20M/R/V, 33F, 35G, 36I, 43T, 46L, 47V, 54A/M/V, 58E, 69K, 74P, 82L/T, 83D y 84V. Los aislados clínicos que mostraron una disminución ≥10 en la susceptibilidad al APTIVUS® incluyeron las ocho o más mutaciones asociadas con APTIVUS®. En los estudios clínicos de fases II y III, 276 pacientes con genotipos bajo tratamiento han demostrado que las mutaciones predominantes que surgen con el tratamiento con APTIVUS® son L33F/I, V82T/L y I84V. Se requiere generalmente la combinación de estos tres tipos para una susceptibilidad reducida. Las mutaciones en la posición 82 se presentan por medio de dos rutas: una a partir de la mutación preexistente de 82A seleccionando a 82T; la otra, del tipo salvaje 82V, seleccionando a 82L. En un estudio con pacientes vírgenes a tratamiento, fue investigado el desarrollo de la resistencia a proteasas en los pacientes que experimentaban rebote virológico después de un régimen que contenía la administración de APTIVUS®, coadministrado con ritonavir a dosis bajas. De los diecisiete pacientes evaluados con el virus sin mutaciones preexistentes a inhibidores de la proteasa, en ninguno se desarrolló resistencia al inhibidor de la proteasa. Resistencia cruzada: El APTIVUS® mantiene una importante actividad antiviral (resistencia < 4 veces) contra la mayoría de los aislados clínicos de VIH-1 que muestra una disminución en la susceptibilidad postratamiento a los inhibidores de la proteasa actualmente aprobados: amprenavir, atazanavir, indinavir, lopinavir, ritonavir, nelfinavir y saquinavir. No es común una resistencia mayor a 10 veces al APTIVUS® ( < 2,5% de los aislados analizados) en virus obtenidos de pacientes que han estado bajo diversos tratamientos y que ha recibido múltiples inhibidores peptídicos de la proteasa. Los virus resistentes al APTIVUS® que surgen in vitro del VIH-1 tipo salvaje muestran una disminución en la susceptibilidad a los inhibidores de la proteasa, amprenavir, atazanavir, indinavir, lopinavir, nelfinavir y ritonavir, pero permanecen sensibles a saquinavir. Farmacocinética: con el objeto de alcanzar concentraciones efectivas de APTIVUS® en plasma y un régimen de dosificación de dos veces al día, resulta esencial la coadministración con APTIVUS®, coadministrado con ritonavir a dosis bajas. El ritonavir actúa inhibiendo el citocromo hepático P450 CYP3A, la bomba de flujo intestinal de glicoproteína P (P-gp) y posiblemente el citocromo intestinal P450 CYP3A. Como se demostró en una evaluación de un rango de dosis en 113 voluntarios sanos, varones y mujeres VIH negativos, el ritonavir incrementa el área bajo la curva (ABC0-12h), la concentración máxima (Cmáx) y la concentración mínima (Cmín) y disminuye la depuración del APTIVUS®. APTIVUS®, coadministrado con ritonavir a dosis bajas (500 mg/200 mg administrado dos veces al día), se asoció con un incremento de 29 veces la media geométrica en el estado estable matutino a través de concentraciones de plasma comparado con una dosis de APTIVUS® de 500 mg administrado dos veces al día sin ritonavir. Un estudio de pacientes infectados con VIH evaluó la farmacocinética y seguridad de APTIVUS® coadministrado con ritonavir a dosis bajas 500 mg/200 mg administrado con y sin lopinavir, amprenavir o saquinavir comparado con ritonavir 100 mg administrado con lopinavir, amprenavir o saquinavir. La concentración media sistémica de ritonavir cuando 200 mg de ritonavir se dieron con APTIVUS® fue similar a las concentraciones observadas cuando se dieron 100 mg con los otros inhibidores de proteasa. Absorción: la absorción del APTIVUS® en humanos es limitada, aunque la cuantificación absoluta de la absorción aún no se encuentra disponible. APTIVUS® es un sustrato P-gp. Las concentraciones máximas en plasma se alcanzan en el intervalo de 1 a 5 horas después de la administración, dependiendo de la dosis utilizada. Con la dosificación repetida, las concentraciones en plasma de APTIVUS® son menores de lo esperado a partir de los datos de la dosis única, probablemente debido a la inducción y transporte de la enzima hepática. El estado estable se logra en la mayoría de los pacientes después de 7 días de dosificación. El APTIVUS®, coadministrado con ritonavir a dosis bajas, presenta una farmacocinética lineal en el estado estable. Administrando 500 mg de APTIVUS® concomitante con 200 mg de ritonavir 2 veces al día durante 2 a 4 semanas y sin restricción de comida, produce un pico promedio en la concentración en plasma de APTIVUS® (Cmáx) de 94,8 ± 22,8 mm para pacientes femeninos (n=14) y 77,6 ± 16,6 mm para pacientes masculinos (n=106), ocurre aproximadamente 3 horas después de la administración. El promedio en estado estable bajo concentración preliminar a la dosis matutina fue 41,6 ± 24,3 mm para pacientes femeninos y 35,6 ± 16,7 mm para pacientes masculinos. El ABC de APTIVUS® encima a las 12 horas de administrado promedió intervalos de 851 ± 309 mm.h (CL = 1,15 l/h) en pacientes femeninos y 710 ± 207 mm.h (CL = 1,27 l/h) en pacientes masculinos. El promedio de vida media fue 5,5 (mujeres) y 6,0 horas (hombres). Efectos del alimento en la absorción oral: el APTIVUS® puede administrarse en forma segura con alimentos estándar o altos en grasas. Las cápsulas de APTIVUS®, coadministrado con ritonavir a dosis bajas, deberán ser deglutidas junto con el alimento. Se analizaron las cápsulas de APTIVUS® administradas en condiciones de un alimento alto en grasas o con un ligero refrigerio de pan tostado y licuado en un estudio de dosis múltiple. El alimento aumentó la biodisponibilidad (estimación del punto ABC 1,31, el intervalo de confianza 1,23 - 1,39), pero tuvo un efecto mínimo sobre las concentraciones máximas de APTIVUS® (Cmáx estimado del punto 1,16, intervalo de confianza 1,09 - 1,24). Para las cápsulas de APTIVUS® coadministradas con ritonavir en estado estable, el alimento no afecta las concentraciones de tipranavir comparado con el estado de ayuno. Sin embargo, el ritonavir debe administrarse con alimento para mejorar su tolerancia. Cuando se coadministró APTIVUS® con dosis bajas de ritonavir y 20 ml de un antiácido líquido a base de aluminio y magnesio, el ABC12h, la Cmáx y la concentración plasmática a las 12 horas (C12h) del APTIVUS® se redujeron en un rango de 25-29%. Debe considerarse la separación de la administración de APTIVUS®, coadministrado con ritonavir a dosis bajas cuando se administre concomitantemente con un antiácido esto, con el objetivo de prevenir una reducción en la absorción del APTIVUS®. Distribución: el APTIVUS® se une de manera importante a las proteínas plasmáticas ( > 99,9%). A partir de las muestras clínicas de voluntarios sanos y en los pacientes VIH positivos que recibieron APTIVUS® sin ritonavir, se detectó que la fracción media de APTIVUS® no unida a las proteínas plasmáticas fue semejante en ambas poblaciones (voluntarios sanos 0,015% ± 0,006%; pacientes VIH positivos 0.019% ± 0.076%). Las concentraciones totales de APTIVUS® en plasma para estas muestras variaron de 9 a 82 mm. La fracción no unida de APTIVUS® pareció ser independiente de la concentración total del fármaco en este rango de concentración. No se han realizado estudios para determinar la distribución de APTIVUS® en el líquido cerebroespinal o en el semen humano. Metabolismo: los estudios de metabolismo in vitro con microsomas hepáticos humanos indicaron que el CYP3A4 es la isoforma CYP predominante involucrada en el metabolismo de APTIVUS®. La depuración de APTIVUS® administrado por vía oral disminuyó después de la adicción de ritonavir, lo cual puede representar una eliminación disminuida de primer paso del fármaco en el tracto gastrointestinal, así como en el hígado. El metabolismo del APTIVUS® en presencia de dosis bajas de ritonavir es mínimo. En un estudio en humanos con APTIVUS® marcado con 14C (APTIVUS® marcado con 14C/ritonavir, 500 mg/200 mg administrado dos veces al día), predominó el APTIVUS® inalterado y representó un 98,4% o más de la radioactividad total en plasma, circulando a las 3, 8 o 12 horas después de la dosificación. Solamente se encontraron pocos metabolitos en el plasma, y todos a nivel de trazas (0,2% o menos de la radioactividad en plasma). En las heces, el APTIVUS® inalterado representó la mayoría de la radioactividad fecal (79,9% de la radioactividad fecal). El metabolito fecal más abundante, con 4,9% de la radioactividad fecal (3,2% de la dosis), fue el metabolito hidroxilo de APTIVUS®. En la orina, se encontró APTIVUS® inalterado a nivel de trazas (0,5% de la radioactividad en orina). El metabolito urinario más abundante, con 11,0% de la radioactividad en orina (0,5% de la dosis) fue el conjugado glucurónido del APTIVUS®. Eliminación: la administración de APTIVUS® marcado con 14C en pacientes (n=8) que recibieron APTIVUS®/ritonavir/r 500 mg/200 mg dos veces al día dosificados hasta el estado estable demostró que la mayoría de la radioactividad (mediana 82,3%) se excretó en las heces, mientras que solamente una mediana de 4,4% de la dosis radioactiva administrada se recuperó en la orina. Además, la mayoría de la radioactividad (56,3%) se excretó entre las 24 y las 96 horas posterior a la dosificación. La vida media efectiva de eliminación promedio de APTIVUS®, coadministrado con ritonavir a dosis bajas en voluntarios sanos (n=67) y en pacientes adultos infectados con VIH (n=120), fue de 4,8 y 6,0 horas, respectivamente, en estado estable después de una dosis de 500 mg/200 mg dos veces al día con una comida ligera. Poblaciones especiales: diferencias farmacocinéticas relacionadas con la edad: la evaluación de las concentraciones de APTIVUS® en plasma en estado estable a las 10-14 horas después de la dosificación a partir de los estudios RESIST-1 y RESIST-2 demostró que no existió cambio en la mediana de las concentraciones mínimas de APTIVUS® a medida que la edad se incrementaba para cualquier sexo hasta los 65 años de edad. No existió un número suficiente de mujeres mayores en ambos estudios; sin embargo, se observó una tendencia consistente de que las concentraciones de APTIVUS® se incrementan con la edad hasta los 80 años en los hombres. Diferencias farmacocinéticas relacionadas con el género: la evaluación de las concentraciones plasmáticas de APTIVUS® en el estado estable a las 10-14 horas después de la dosificación en los estudios RESIST-1 y RESIST-2 demostró que las mujeres presentaron generalmente una mayor concentración de APTIVUS® que los hombres. Después de 4 semanas de administrar APTIVUS®/ritonavir 500 mg/200 mg dos veces al día, la concentración mediana de APTIVUS® en plasma fue de 43,9 mm para mujeres y 31, mM para hombres. Esta diferencia en las concentraciones no justifica un ajuste en la dosis. Diferencias farmacocinéticas relacionadas con la raza: la evaluación de las concentraciones plasmáticas de APTIVUS® en el estado estable a las 10-14 horas después de la dosificación en los estudios RESIST-1 y RESIST-2 demostró que los hombres de raza blanca presentaron generalmente una mayor variación en las concentraciones de APTIVUS® que los hombres de raza negra, pero la concentración mediana y el rango que muestra la mayoría de los datos son comparables entre las razas. Las mujeres de cualquier raza tuvieron generalmente concentraciones mayores de APTIVUS® que los hombres. Insuficiencia renal: la farmacocinética del APTIVUS® no se ha estudiado en pacientes con insuficiencia renal. Sin embargo, debido a que la depuración renal del APTIVUS® es despreciable, no se espera una disminución de la depuración total en pacientes con insuficiencia renal. Insuficiencia hepática: en un estudio que comparó 9 pacientes con insuficiencia hepática leve (escala Child-Pugh A) con 9 controles, la disposición de la farmacocinética de la dosis única y la dosis múltiple de APTIVUS® y ritonavir aumentó en pacientes con insuficiencia hepática, pero aún dentro del rango observado en los estudios clínicos. No se requiere de un ajuste en la dosis en pacientes con insuficiencia hepática leve. No se ha evaluado aún la influencia de la insuficiencia hepática moderada (escala Child Pugh B) en la farmacocinética multidosis del APTIVUS® o ritonavir. El APTIVUS® debe contraindicarse en aquellos con insuficiencia hepática moderada o severa (ver Contraindicaciones). Descripción de los estudios clínicos: pacientes adultos con tratamiento experimentado: estudios RESIST-1 y RESIST-2: APTIVUS®, coadministrado con ritonavir a dosis bajas 500 mg/200 mg administrado dos veces al día + régimen optimizado de base (OBR, por sus siglas en inglés) vs. comparador IP/Ritonavir dos veces al día + OBR): los siguientes datos clínicos se derivan de los análisis de los datos a 48 semanas de los estudios que se están realizando actualmente (RESIST-1 y RESIST-2) midiendo los efectos sobre los niveles de ARN del VIH-1 en el plasma y los conteos de células CD4. A la fecha, no existen resultados de los estudios controlados que evalúen el efecto del APTIVUS® en la progresión clínica del VIH. RESIST-1 y RESIST-2 son estudios en proceso, con distribución al azar, abiertos, multicéntricos, que involucran pacientes VIH-positivos, los cuales han recibido terapia de las 3 clases de antirretrovirales. Estos estudios evalúan el tratamiento con APTIVUS® coadministrado con ritonavir a dosis bajas (APTIVUS®/ritnavir) más un OBR definido individualmente para cada paciente con base en las pruebas de resistencia genotípica y antecedentes del paciente. El régimen de comparación incluyó a un inhibidor de proteasa (IP) reforzado con ritonavir (CPI/r, también definido individualmente) más un OBR. El IP reforzado con ritonavir fue elegido de entre saquinavir, amprenavir, indinavir o lopinavir/ritonavir. Todos los pacientes habían recibido por lo menos dos regímenes antirretrovirales basados en un IP y estaban fallando a un régimen basado en un IP al momento de entrar en el estudio. Por lo menos una mutación primaria del gen de proteasa entre 30N, 46I, 46L, 48V, 50V, 82A, 82F, 82L, 82T, 84V o 90M tuvo que estar presente al inicio del estudio, con no más de dos mutaciones en los codones 33, 82, 84 o 90. Después de 8 semanas, los pacientes del brazo comparador, quienes reunían el criterio definido del protocolo de falta de una respuesta virológica inicial, tuvieron la opción de discontinuar el tratamiento y cambiar a APTIVUS®/ritonavir en un estudio separado de continuación. Hubo 1.483 pacientes (APTIVUS®/ritonavir: n=746, CPI/ritonavir: n=737) incluidos en el análisis primario de los estudios RESIST combinados. Los grupos de pacientes tenían medianas de edad de 43 años (intervalo 17-80 años) y 42 años (mediana 21-72 años) para APTIVUS®/ritonavir y CPI/ritonavir, respectivamente. Los pacientes fueron 84% y 88% hombres, 77% y 74% de raza blanca, 12,6% y 13,3% de raza negra y 0,7% y 1,2% asiáticos para los grupos APTIVUS®/ritonavir y CPI/ritonavir, respectivamente. En los brazos de APTIVUS® y del comparador, la mediana de conteos de células CD4 al inicio del tratamiento fue de 158 y 166 células/mm3, respectivamente, (intervalos intercuartilares (IIC) 66-285 y 53-280 células/mm3): la mediana de concentraciones plasmáticas de ARN del VIH-1 al inicio del estudio fueron de 4,79 y 4.80 log10 copias/ml, respectivamente (IIC 4,32-5,24 y 4,25-5,27 log10 copias/ml). La respuesta y los resultados del tratamiento al azar en la semana 24 se presentan en la tabla siguiente.
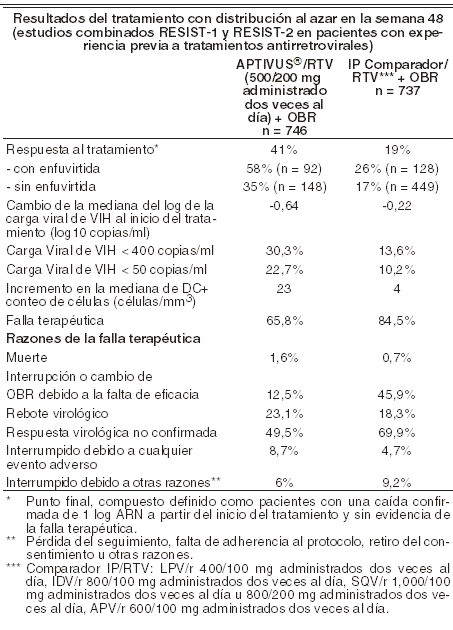
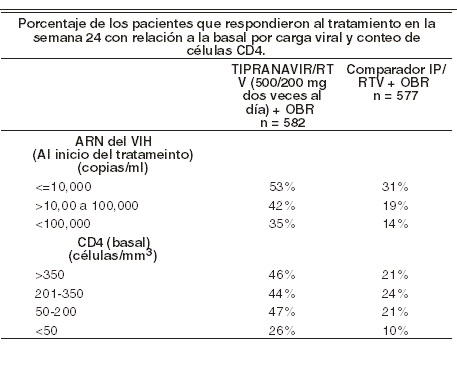
Los datos de los estudios RESIST demostraron también que APTIVUS®, coadministrado con ritonavir a dosis bajas, exhibe una mejor respuesta al tratamiento a las 48 semanas cuando el OBR contiene agentes antirretrovirales genotípicamente disponibles (por ejemplo, enfuvirtida). A lo largo de las 96 semanas de tratamiento, la mediana de tiempo al fracaso del tratamiento fue de 115 días entre los pacientes tratados con APTIVUS®/ritonavir y de 0 días entre los pacientes tratados con CPI/ritonavir. En los pacientes que recibieron nuevo enfuvirtida (definido como inicio de enfuvirtida por vez primera), la mediana de tiempo de fracaso al tratamiento fue de 587 días entre los pacientes tratados con APTIVUS®/ritonavir y de 60 días entre los pacientes tratados con CPI/ritonavir. Análisis de resistencia a tipranavir en pacientes con experiencia a tratamientos antirretrovirales: las tasas de respuesta a APTIVUS® coadministrado con dosis bajas de ritonavir se evaluaron por genotipo y fenotipo basal al tipranavir. Se evaluaron las relaciones entre la susceptibilidad fenotípica al tipranavir, las mutaciones asociadas a resistencia al tipranavir y la respuesta al tratamiento con APTIVUS® coadministrado con dosis bajas de ritonavir. Mutaciones asociadas a resistencia al tipranavir: se han evaluado las respuesta virológica y al tratamiento a la terapia con APTIVUS® coadministrado con dosis bajas de ritonavir usando una calificación de mutación asociada al tipranavir basada en el genotipo basal en los pacientes de los estudios RESIST-1 y RESIST-2. Esta calificación (cuenta los 16 aminoácidos que se han asociado con susceptibilidad disminuida a tipranavir y/o menor respuesta de la carga viral: 10V, 13V, 20M/R/V, 33F, 35G, 36I, 43T, 46L, 47V, 54A/M/V, 58E, 69K, 74P, 82L/T, 83D y 84V) se aplicó a las secuencias de proteasa viral basales. Se estableció una correlación entre la calificación de mutación al tipranavir y la respuesta al tratamiento con APTIVUS® coadministrado con dosis bajas de ritonavir a las semanas 2 y 48. A la semana 48, una mayor proporción de pacientes que recibían APTIVUS® coadministrado con dosis bajas de ritonavir alcanzó una respuesta al tratamiento en comparación con el CPI/ritonavir para casi todas las posibles combinaciones de mutaciones de resistencia genotípica (Tabla 1).
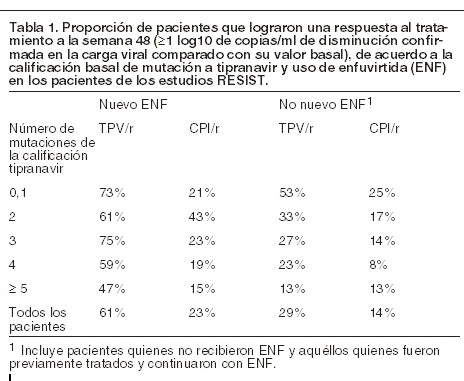
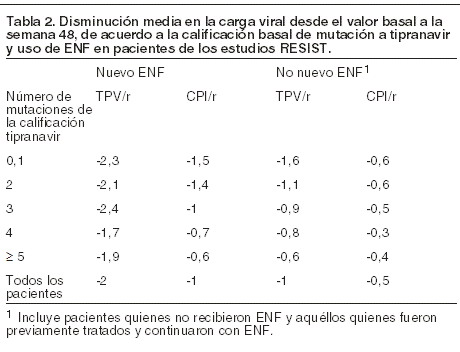
Las reducciones sostenidas del ARN del VIH-1 hasta la semana 48 (Tabla 2) se observaron principalmente en pacientes quienes recibieron APTIVUS® coadministrado con dosis bajas de ritonavir y nuevo ENF. Si los pacientes no recibieron APTIVUS® coadministrado con dosis bajas de ritonavir con nuevo ENF, se observaron respuestas al tratamiento reducidas con respecto al uso de nuevo ENF.
Mutaciones de proteasa en las posiciones 33, 82, 84 y 90: las mutaciones en dos, tres o más de estas posiciones resultó en menor susceptibilidad al APTIVUS® coadministrado con dosis bajas de ritonavir y cuatro mutaciones resultaron en resistencia. Resistencia fenotípica a tipranavir: el cambio al doble fenotípico basal al tipranavir en los aislamientos se correlacionó con menor respuesta viral. Los aislamientos con cambio de 0 a 3 veces a partir de la basal se consideraron susceptibles; aislamientos con > 3 a 10 veces tuvieron menor susceptibilidad; aislamientos con > 10 veces son resistentes. Las conclusiones respecto de la relevancia de las mutaciones particulares o patrones mutacionales están sujetos a cambios con los datos adicionales, y se recomienda siempre consultar con los sistemas de interpretación actuales para los resultados de las pruebas de análisis de resistencia.
Contraindicaciones: Hipersensibilidad al principio activo o a cualquiera de los componentes del producto. El uso del producto está contraindicado en el caso de condiciones hereditarias raras que pudieran ser incompatibles con un excipiente del producto (por favor, refiérase a Precauciones generales). APTIVUS® está contraindicado en pacientes con insuficiencia hepática moderada ó severa (escala de Chil-Pugh Clase B ó C). La coadministración de APTIVUS® y dosis bajas de ritonavir está contraindicada con fármacos que son altamente dependientes del CYP3A para su depuración y los cuales pueden elevar las concentraciones en plasma, estando estas asociadas con eventos serios y/o amenazadores para la vida. Estos fármacos incluyen antiarrítmicos (amiodarona, bepridil, flecainida, propafenona y quinidina), antihistamínicos (astemizol y terfenadina), derivados de la ergotamina (dihidroergotamina, ergonovina, ergotamina y metilergonovina), agentes de motilidad gastrointestinal (GI) (cisaprida), neurolépticos (pimozida) y sedantes/hipnóticos (midazolam y triazolam administrados por vía oral).
Precauciones generales: APTIVUS® debe administrarse con dosis bajas de ritonavir para asegurar su efecto terapéutico (veáse Dosis y vía de administración). El no observar la administración correcta de APTIVUS® con ritonavir resultará en una reducción de los niveles en plasma de APTIVUS®, que pueden ser insuficientes para alcanzar el efecto antiviral deseado. Debe instruirse adecuadamente a los pacientes sobre la correcta administración de los medicamentos utilizados. APTIVUS® no es un medicamento que cure la infección de VIH-1 o SIDA. Los pacientes que reciben APTIVUS® o cualquier otra terapia antirretroviral pueden continuar desarrollando las infecciones oportunistas y otras complicaciones de la infección por VIH-1. No se ha demostrado que la terapia con APTIVUS® reduzca el riesgo de transmisión de VIH-1 a otras personas. Los estudios clínicos de APTIVUS® no incluyeron números suficientes de pacientes de 65 años y mayores para determinar si responden de forma diferente a los sujetos mas jóvenes. En general, debe tenerse precaución en la administración y el monitoreo de APTIVUS® en pacientes mayores que reflejan una mayor frecuencia en la disminución de la función hepática, renal o cardíaca y de una enfermedad concomitante u otras terapias con fármacos. El uso del producto está contraindicado en el caso de condiciones hereditarias raras que pudieran ser incompatibles con un excipiente del producto. Insuficiencia y toxicidad hepática: APTIVUS®, coadministrado con ritonavir a dosis bajas, ha sido asociado con reportes de hepatitis clínica y descompensación hepática, incluyendo algunas muertes. APTIVUS® debe ser usado con precaución en pacientes coinfectados con hepatitis B o C, y aumentar el monitoreo clínico y de laboratorio. Deben realizarse las pruebas de laboratorio apropiadas antes de iniciar y durante el tratamiento con APTIVUS® coadministrado con ritonavir a dosis bajas. Debe considerarse un incremento del monitoreo de la función hepática cuando se administran APTIVUS® con dosis bajas de ritonavir en pacientes con niveles elevados de ALT y AST al inicio del tratamiento, o con hepatitis activa B o C, los pacientes con hepatitis B o C subyacente o marcadas elevaciones en las transaminasas antes del tratamiento pueden tener un mayor riesgo de desarrollar un incremento adicional de transaminasas o una descompensación hepática. Si existieran elevaciones asintomáticas en los niveles de AST o ALT mayores de 10X ULN, deberá suspenderse el tratamiento con APTIVUS®. Si además se identifican otras causas de la elevación (p.ej. hepatitis aguda por virus A, B o C, enfermedades de la vesícula biliar, otros medicamentos) o si el beneficio potencial pesa más que el riesgo, entonces se puede considerar retornar al tratamiento con APTIVUS® cuando los niveles de AST/ALT hayan regresado a los niveles basales. Si se presentan datos de hepatitis sintomática, deberá suspenderse el tratamiento con APTIVUS®. Si además se identifican otras causas de la elevación (ej. hepatitis aguda por virus A, B ó C, enfermedades de la vesícula biliar, otros medicamentos) o si el beneficio potencial pesa más que el riesgo, entonces se puede considerar retornar al tratamiento con APTIVUS® cuando los niveles de AST/ALT hayan regresado a los niveles basales. APTIVUS®, coadministrado con ritonavir a dosis bajas, ha sido asociado con reportes de hepatitis clínica y descompensación hepática, incluyendo algunas muertes. Esto ha ocurrido generalmente en pacientes con enfermedad avanzada por VIH que se encuentran bajo tratamiento con múltiples medicamentos concomitantes. Una interrelación causal de APTIVUS®, coadministrado con ritonavir a dosis bajas, no pudo ser establecida. Pacientes con signos o síntomas de hepatitis deberán discontinuar el tratamiento APTIVUS® coadministrado con ritonavir a dosis bajas y buscar evaluación médica. Deberá tener precaución cuando administre APTIVUS® a pacientes con anormalidades en las enzimas del hígado o antecedente de hepatitis. APTIVUS® se metaboliza principalmente por el hígado. Por lo tanto, debe tenerse precaución cuando se administre este fármaco a pacientes con insuficiencia hepática debido a que pueden incrementarse las concentraciones de APTIVUS®. APTIVUS® está contraindicado en pacientes con insuficiencia hepática moderada o severa (escala Child-Pugh-Turcotte Clase B o C). Para mayor información sobre la farmacocinética de dosis múltiples de APTIVUS® en pacientes con insuficiencia hepática véase, Farmacocinética en pacientes adultos. Pacientes vírgenes a tratamiento: en un estudio realizado en pacientes vírgenes a tratamiento, el 16,2% experimentan un grado 3 o 4 de elevación en los niveles de ALT cuando recibieron APTIVUS®, coadministrado con ritonavir a dosis bajas 500 mg/200 mg en la semana 48 del tratamiento, por lo que no se recomienda usar el tratamiento de APTIVUS®, coadministrado con ritonavir a dosis bajas, en pacientes vírgenes infectados con el virus de tipo primitivo. Daño renal: debido a que la depuración renal del APTIVUS® es despreciable, no se espera un incremento de las concentraciones plasmáticas en pacientes con insuficiencia renal. Hemofilia: se han presentado reportes de incremento en el sangrado, incluyendo hematomas espontáneos en la piel y hemartrosis en pacientes con hemofilia tipo A y B bajo tratamiento con inhibidores de la proteasa. En algunos pacientes, adicionalmente se administró el factor VIII. En más de la mitad de los casos reportados, se continúo el tratamiento con inhibidores de la proteasa y se reintrodujo si el tratamiento se había interrumpido. No ha sido establecida una relación causal entre los inhibidores de la proteasa y estos eventos. Hemorragia intracraneal: APTIVUS®, coadministrado con ritonavir a dosis bajas, ha sido asociado con casos de hemorragia intracraneal (HIC) fatal y no fatal en algunos pacientes, muchos de los cuales presentaban otras condiciones médicas asociadas o bien estaban recibiendo medicamentos concomitantes, los cuales pudieron haber causado o contribuido a la presentación de dichos eventos. En general, no ha sido observada ninguna pauta de existencia de parámetros hematológicos o de coagulación anormales, ni tampoco previos al desarrollo de hemorragia intracraneal. Previamente, se había observado un incremento en el riesgo de hemorragia intracraneal en pacientes con enfermedad por VIH/SIDA avanzada en pacientes tratados con APTIVUS® en estudios clínicos. No se ha establecido una relación directa entre APTIVUS® y hemorragia intracraneal. Agregación plaquetaria y coagulación: APTIVUS®, coadministrado con ritonavir a dosis bajas, deberá ser utilizado con precaución en pacientes en quienes exista riesgo incrementado de sangrado por trauma, cirugía u otras condiciones médicas, o quienes han recibido medicamentos que se conoce incrementan el riesgo de sangrado tales como agentes antiplaquetarios y anticoagulantes, o en quienes reciben dosis altas suplementarias de vitamina E. En experimentos in vitro, se observó que APTIVUS® inhibe la agregación plaquetaria humana a niveles que se presentan durante la exposición de pacientes que reciben la combinación APTIVUS® coadministrado con ritonavir a dosis bajas. En ratas, la coadministración de vitamina E aumentó los efectos hemorrágicos del tipranavir (ver Toxicología). Sin embargo, el análisis del plasma almacenado de adultos tratados con cápsulas de APTIVUS® más ritonavir a dosis bajas han demostrado que no hubo un efecto del tipranavir sobre los factores de coagulación dependientes de vitamina K (factores II y VII), el factor V o sobre los tiempos de protrombina o tromboplastina parcial activada. Diabetes mellitus/hiperglicemia: se han reportado nuevos casos o exacerbación de diabetes mellitus preexistente e hiperglicemia durante la vigilancia postcomercialización en pacientes infectados con VIH que recibían terapia con un inhibidor de la proteasa. Algunos pacientes requirieron iniciar el uso, o bien, llevar a cabo un ajuste en la dosis de insulina o agentes hipoglicémicos orales para el tratamiento de estos eventos. En algunos casos, se presentó cetoacidosis diabética. En estos pacientes, se discontinúo la terapia con inhibidor de la proteasa, pero la hiperglucemia persistió en algunos casos. Debido a que estos eventos se han reportado voluntariamente durante la práctica clínica, no se pueden realizar estimados de la frecuencia. No se ha establecido la relación causal entre la terapia con un inhibidor de proteasa y estos eventos. Incremento en los lípidos: el tratamiento con APTIVUS®, coadministrado con ritonavir a dosis bajas, y otros agentes antirretrovirales, ha resultado en un incremento de los triglicéridos totales y colesterol en el plasma. Las pruebas de triglicéridos y colesterol deben realizarse antes de iniciar la terapia con APTIVUS® y durante ésta. El aumento de lípidos relacionados con el tratamiento debe manejarse como se considere clínicamente apropiado. Redistribución de grasas: se ha asociado la terapia de combinación antirretroviral con la redistribución de grasas en el cuerpo (lipodistrofia) en pacientes infectados con VIH. Actualmente se desconocen las consecuencias de estos eventos a largo plazo. El conocimiento del mecanismo es incompleto. Se ha elaborado la hipótesis de una relación entre la lipomatosis visceral y los inhibidores de la proteasa, así como en la lipoatrofia y los nucleósidos inhibidores de la transcriptasa inversa (NITR). Se ha asociado un mayor riesgo de lipodistrofia con factores individuales como una mayor edad, y con factores relacionados con el fármaco tales como una mayor duración del tratamiento antirretroviral y alteraciones metabólicas asociadas. El examen clínico debe incluir la evaluación de los signos físicos de la redistribución de grasa. Debe considerarse la cuantificación de lípidos en suero y glucosa en sangre en ayunas. Las alteraciones de los lípidos deben manejarse como se considere clínicamente apropiado. Síndrome de reconstitución inmune: se ha reportado el síndrome de reconstitución inmune en pacientes tratados con terapia de combinación antirretroviral, incluyendo APTIVUS®. Durante la fase inicial del tratamiento de combinación antirretroviral, los pacientes cuyo sistema inmune responde puede desarrollar una respuesta inflamatoria a infecciones oportunistas indolentes o residuales (como infección por Mycobacterium avium, citomegalovirus, neumonía por Pneumocystis carinii, tuberculosis o reactivación de herpes simple y herpes zóster), los cuales pueden necesitar de una evaluación y tratamiento posterior. Advertencias sobre el uso concomitante con otros fármacos: APTIVUS®, coadministrado con ritonavir a dosis bajas puede cambiar la exposición de otros fármacos en plasma y otros fármacos pueden alterar la exposición de APTIVUS® y ritonavir en plasma. Para una descripción de los mecanismos y los mecanismos potenciales que contribuyen al perfil de interacción de APTIVUS®, ver la sección de Interacciones medicamentosas y de otro genero. Inhibidores de proteasa: el uso concomitante de APTIVUS® coadministrado con ritonavir a dosis bajas, con los inhibidores de proteasa (coadministrados con ritonavir a dosis bajas) resulta en disminuciones significativas en las concentraciones plasmáticas de estos inhibidores de proteasa. No se recomienda combinar inhibidores de proteasa con APTIVUS® coadministrado con ritonavir a dosis bajas. Los pacientes que reciben la combinación de APTIVUS®/amprenavir, o ambos coadministrados con ritonavir a dosis bajas, pueden tener un incremento en el riesgo de presentar elevaciones de las transaminasas hepáticas Grado 3/4. Inhibidores de HMG-CoA reductasa: los inhibidores de HMG-CoA reductasa, simvastatina y lovastatina son altamente dependientes del CYP3A4 para su metabolismo, por lo cual no se recomienda el uso concomitante de APTIVUS® coadministrado con ritonavir a dosis bajas con estos medicamentos debido a un incremento en el riesgo de miopatía, incluyendo rabdomiólisis. También debe tenerse precaución y considerarse el uso de las mínimas dosis posibles de atorvastatina si APTIVUS®, coadministrado con ritonavir a dosis bajas, se utiliza junto con la atorvastatina, la cual es metabolizada en un menor grado por CYP3A4. Se debe considerar el uso de otros inhibidores de HMG-CoA reductasa que no sean atorvastatina. Inhibidores de fosfodiesterasa (PDE5, por sus siglas en inglés): debe tenerse particular precaución cuando se prescriban inhibidores de fosfodiesterasa (PDE5) (p.ej. sildenafil, vardenafil o tadalafil) en pacientes que reciben APTIVUS® coadministrado con ritonavir a dosis bajas. Se espera que la coadministración de APTIVUS® y ritonavir a dosis bajas con inhibidores de PDE5 incremente substancialmente las concentraciones de los inhibidores de PDE5 y pueda resultar en un aumento en los eventos adversos asociados con el inhibidor de PDE5, incluyendo hipotensión, alteraciones de la visión y priapismo. El uso concomitante de APTIVUS®, coadministrado con ritonavir a dosis bajas, con tadalafil resultó en un aumento en la exposición al tadalafil con la primera dosis de APTIVUS® coadministrado con ritonavir a dosis bajas y ningún cambio en la ex
posición al tadalafil con el APTIVUS®/ritonavir en estado estable. Si se usa tadalafil dentro de los primeros días de tratamiento de APTIVUS®/ritonavir, debe administrarse la dosis más baja de tadalafil. Después de 7 a 10 días de administrar APTIVUS®/ritonavir, puede aumentarse la dosis de tadalafil hasta donde sea clínicamente necesario (ver Interacciones medicamentosas y de otro género). Anticonceptivos orales y estrógenos: debido a que los niveles de etinil estradiol se reducen, deben utilizarse medidas anticonceptivas alternas o adicionales cuando los anticonceptivos administrados por vía oral basados en estrógenos se administran concomitantemente con APTIVUS® coadministrado con ritonavir a dosis bajas. Los pacientes que usan estrógenos como terapia de hormonas de replazo deberán ser clínicamente monitoreados sus signos de deficiencia de estrógenos. Las mujeres que utilizan estrógenos pueden tener un incremento en el riesgo de presentar erupción cutánea leve. Analgésicos narcóticos: la coadministración de APTIVUS® y dosis bajas de ritonavir con una dosis única de metadona resulta en la reducción del 50% aproximadamente en las concentraciones de esta última (ABC y Cmáx). Por lo tanto, en estos casos, debe monitorizarse a los pacientes para determinar si se presenta el síndrome de abstinencia opiácea. Por lo anterior, puede ser necesario incrementar la dosis de metadona. Se espera que APTIVUS®, coadministrado con ritonavir a dosis bajas, disminuya las concentraciones de meperidina e incremente las concentraciones del metabolito normeperidina. El aumento en la dosificación y el uso a largo plazo de meperidina con APTIVUS®, coadministrado con ritonavir a dosis bajas, no se recomiendan debido a un incremento en las concentraciones del metabolito anteriormente mencionado, el cual posee una actividad analgésica y también actúa estimulando la actividad del sistema nervioso central (SNC) (por ej., convulsiones). Anticonvulsivantes: debe tenerse cuidado cuando se prescriban carbamazepina, fenobarbital o fenitoína. APTIVUS® puede ser menos efectivo debido a la reducción en la concentración plasmática de tipranavir en pacientes que toman estos fármacos concomitantemente. Disulfiram/metronidazol: las cápsulas suaves de gelatina de APTIVUS® contienen alcohol el cual puede generar reacciones relacionadas al disulfiram cuando se coadministra con disulfiram u otros fármacos que producen esta reacción (p.ej., metronidazol). Rifampicina (rifampin): no se recomienda el uso concomitante de APTIVUS® y rifampicina. Se espera que la coadministración de los inhibidores de proteasa, incluyendo APTIVUS®, con rifampicina, disminuya substancialmente las concentraciones del inhibidor de proteasa y pueda resultar en niveles subóptimos de APTIVUS®, conduciendo a una pérdida de la respuesta virológica y una posible resistencia a este fármaco o a la clase de los inhibidores de la proteasa. Hierba de San Juan (Hypericum perforatum): se espera que la coadministración de los inhibidores de proteasa incluyendo APTIVUS® con la hierba de San Juan disminuyan substancialmente las concentraciones del inhibidor de la proteasa y pueda resultar en niveles subóptimos de APTIVUS® conduciendo con ello a una pérdida de la respuesta virológica y una posible resistencia al APTIVUS® o a la clase de inhibidores de la proteasa por lo que no se recomienda el uso concomitante del tipravanir y la hierba de San Juan (Hypericum perforatum), o productos que la contengan. Propionato de fluticasona: un estudio de interacción del fármaco con sujetos sanos ha mostrado que el ritonavir incrementa significativamente en plasma la exposición de propionato de fluticasona, resultando un decremento significativo de las concentraciones de cortisol sérico. El uso concomitante de APTIVUS®, coadministrado con ritonavir a dosis bajas, y propionato de fluticasona puede producir los mismos efectos. Se han reportado durante el uso postcomercialización efectos sistémicos corticoesteroideos, incluyendo al síndrome de Cushing y supresión adrenal en pacientes que recibieron ritonavir y propionato de fluticasona inhalado o administrado vía intranasal. Por lo tanto, no se recomienda la coadministración de propionato de fluticasona y APTIVUS® coadministrado con ritonavir a dosis bajas, a menos que el beneficio de los pacientes importe más que los riesgos de efectos adversos del corticoesteroide sistémico. Midazolam: si el APTIVUS®/ritonavir se coadministra con midazolam parenteral, debe vigilarse estrechamente al paciente en búsqueda de depresión respiratoria y/o sedación prolongada y debe realizarse un ajuste en la dosificación si se considera pertinente (ver Interacciones medicamentosas y de tro género). Trazadona: el uso concomitante de trazadona y APTIVUS®, coadministrado con ritonavir a dosis bajas, puede incrementar las concentraciones de trazadona en plasma. Se han observado efectos adversos como náusea, vértigo, hipotensión y síncope seguidos de la coadministración de trazadona y ritonavir. Si es usada trazadona con APTIVUS® coadministrado con ritonavir a dosis bajas la combinación deberá ser usada con precaución y se debe considerar una dosis baja de trazadona. APTIVUS® cápsulas contiene hasta 50,4 mg de sorbitol por dosis diaria máxima recomendada. Los pacientes con la rara condición hereditaria de intolerancia a la fructosa no deben tomar esta medicina. Efectos sobre la habilidad para conducir y operar máquinas: no existen estudios específicos acerca de la habilidad para conducir vehículos y operar maquinaria.
Restricciones de uso durante el embarazo y la lactancia: No existen estudios adecuados y bien controlados en mujeres embarazadas para el tratamiento de la infección por VIH-1. APTIVUS® puede utilizarse durante el embarazo solamente si el beneficio potencial justifica el riesgo potencial para el feto. De acuerdo con las recomendaciones para las madres infectadas con VIH, no deberán alimentar del seno materno a sus hijos con el objetivo de evitar el riesgo de transmisión postnatal de VIH; las madres deben interrumpir la lactancia si están bajo tratamiento con APTIVUS®.
Reacciones secundarias y adversas: Se han realizado estudios clínicos con el APTIVUS® coadministrado con ritonavir a dosis bajas como terapia de combinación, en un total de 6.308 adultos VIH-positivos. De estos pacientes, 1.299 pacientes con tratamiento antirretroviral previo recibieron la dosis de 500 mg/200 mg dos veces al día en los estudios clínicos formales. 909 de estos pacientes, incluyendo 541 en los estudios pivotes de fase III RESIST-1 y RESIST-2, se han tratado por, al menos, 48 semanas. En los estudios RESIST-1 y RESIST-2, en el brazo de APTIVUS®, coadministrado con ritonavir a dosis bajas, los eventos adversos más frecuentes fueron: diarrea, náusea, cefalea, pirexia, vómito, fatiga y abdominalgia. Las tasas de eventos adversos por Kaplan-Meier a las 48 semanas que condujeron a la discontinuación del tratamiento se reportaron en el 13,3% de los pacientes dosificados con APTIVUS® y en el 10,8% de los pacientes en el brazo de comparación. Las siguientes características de seguridad clínica (hepatotoxicidad, hiperlipidemia) se observaron en una frecuencia alta entre pacientes tratados con APTIVUS®, coadministado con ritonavir a dosis bajas, cuando se compararon con el brazo comparador de pacientes en las pruebas RESIST. Hepatotoxicidad: la frecuencia de anormalidades grado 3 o 4 de ALAT y/o ASAT fue alta en pacientes con APTIVUS®/ritonavir comparada con pacientes en el brazo de referencia. Los análisis multivariados mostraron que los valores basales de ALAT y/o ASAT encima de DAIDS grado 1 y una coinfección con hepatitis B o C fueron factores de riesgo para estas elevaciones. Hiperlipidemia: elevaciones grado 3 o 4 de triglicéridos y colesterol ocurrieron más frecuentemente en los brazos de APTIVUS® coadministrado con ritonavir a dosis bajas, comparado con el brazo de referencia. La importancia clínica de estas observaciones no se ha establecido. Las reacciones adversas más frecuentes de cualquier intensidad (Grados 1-4) reportadas en los brazos delAPTIVUS® coadministrado con ritonavir a dosis bajas en los estudios clínicos fase III (n =749) se listan a continuación por la clase de órgano/sistema y la frecuencia de acuerdo con las siguientes categorías: > 1/10, > 1/100 y < 1/10. Metabolismo y alteraciones de la nutrición: > 1/100 - < 1/10: hipertrigliceridemia, hiperlipidemia y anorexia. Alteraciones del sistema nervioso: > 1/100 - < 1/10: cefalea. Alteraciones gastrointestinales: > 1/10: diarrea y náusea. > 1/100 - < 1/10: vómito, flatulencia, distensión abdominal, dolor abdominal, evacuaciones sueltas y dispepsia. Alteraciones de la piel y del tejido celular subcutáneo: > 1/100 - < 1/10: erupción cutánea y prurito. Trastornos generales: > 1/100 - < 1/10: fatiga. Se clasifican a continuación por órgano/sistema y frecuencia de acuerdo con las siguientes categorías, lasreacciones adversas clínicamente significativas de intensidad moderada o severa que se presentaron enmenos del 1% ( < 1/100) de los pacientes adultos en todos los estudios de fases II y III administrados con la dosis de 500 mg/200 mg de APTIVUS®, coadministrado con ritonavir a dosis bajas (n=1397). > 1/1000, < 1/100 y < 1/1000. Alteraciones hemáticas y sistema linfático: > 1/1000 - < 1/100: anemia, neutropenia y trombocitopenia. Alteraciones en el sistema inmune: > 1/1000 - < 1/100: hipersensibilidad. Alteraciones en el metabolismo y la nutrición: > 1/1000 - < 1/100: disminución del apetito, diabetes mellitus, hiperamilasemia e hipercolesterolemia. < 1/1000: deshidratación, hiperglicemia. Alteraciones psiquiátricas: > 1/1000 - < 1/100: insomnio y trastornos en el sueño. Alteraciones del sistema nervioso: > 1/1000 - < 1/100: mareo, neuropatía periférica y somnolencia. < 1/1000: hemorragia intracraneal. Alteraciones respiratorias, torácicas y mediastinales: > 1/1000 - < 1/100: disnea. Alteraciones gastrointestinales: > 1/1000 - < 1/100: enfermedad de reflujo gastroesofágico y pacreatitis. Alteraciones hepatobiliares: > 1/1000 - < 1/100: hepatitis, hepatitis citolítica, hepatitis tóxica y esteatosis hepática. < 1/1000: insuficiencia hepática (incluyendo consecuencias fatales) e hiperbilirrubinemia. Alteraciones en la piel y del tejido subcutáneo: > 1/1000 - < 1/100: exantema, lipoatrofia, lipodistrofia adquirida y lipohipertrofia. Alteraciones del sistema musculoesquelético y del tejido conectivo: > 1/1000 - < 1/100: calambres musculares y mialgia. Alteraciones renales y urinarias: > 1/1000 - < 1/100: insuficiencia renal. Alteraciones generales: > 1/1000 - < 1/100: enfermedad semejante a la influenza, malestar general y pirexia. Pruebas clínicas: > 1/1000 - < 1/100: aumento de las enzimas hepáticas, pruebas de funcionamiento hepático anormales,disminución de peso y aumento de lipasa. Fueron observados en las pruebas RESIST la reactivación de herpes simple e infecciones por virus de varicela-zóster.
Interacciones medicamentosas y de otro género: APTIVUS® es un sustrato, un inductor y un inhibidor del citocromo P450 CYP3A. Sin embargo, cuando se coadministra con el ritonavir a la dosis recomendada, existe una inhibición neta del P450 CYP3A. La coadministración del APTIVUS® coadministrado con ritonavir a dosis bajas con agentes que se metabolizan primariamente por el CYP3A puede resultar en un cambio en las concentraciones plasmáticas de APTIVUS® u otros agentes, lo cual podría alterar sus efectos terapéuticos y los eventos adversos. Los agentes que están específicamente contraindicados debido a la magnitud esperada de la interacción y el potencial de los eventos adversos serios se listan en la sección de Contraindicaciones. Se condujo un estudio de cóctel fenotípico con 16 voluntarios sanos para cuantificar la influencia de la administración de 10 días de administración de cápsulas de APTIVUS®/ritonavir sobre la actividad del CYP 1A2 hepático (cafeína), 2C9 (warfarina), 2C19 (omeprazol), 2D6 (dextrometorfán) y la actividad del CYP3A4/5 intestinal y hepático (midazolam) y la P-glucoproteína (P-gp)(digoxina). Este estudio determinó los efectos de la primera dosis y el estado estable de 500 mg de APTIVUS® coadministrado con 200 mg de ritonavir dos veces al día en forma de cápsula. No hubo un efecto neto sobre el CYP2C9 o sobre el P-gp hepático en la primera dosis o en el estado estable. No hubo un efecto neto después de la primera dosis sobre el CYP1A2, pero hubo una inducción moderada en el estado estable. Hubo una leve inhibición después de la primera dosis sobre el CYP2C19 y una moderada inducción en el estado estable. Se observaron potente inhibición del CYP2D6 y de las actividades del CYP3A4/5 hepático e intestinal después de la primera dosis y en el estado estable. Se inhibió la actividad de la P-gp intestinal después de la primera dosis, perno no hubo un efecto neto en el estado estable. El APTIVUS® se metaboliza por el CYP3A y es un substrato para la P-gp. La coadministración de tipranavir y agentes que inducen al CYP3A y/o P-gp podría disminuir las concentraciones de tipranavir y reducir así su efecto terapéutico. La coadministración de APTIVUS® y productos medicinales que inhiben la P-gp podría aumentar las concentraciones plasmáticas de tipranavir. Inhibidores de la fusión: Enfuvirtida: la coadministración de enfuvirtida con APTIVUS®, coadministrado con ritonavir a dosis bajas se asoció con un incremento en la concentración valle de tipranavir en el estado estable para la población en estudio en aproximadamente 45%. Se han observado aumentos similares también en las concentraciones valle plasmáticas de lopinavir (23%) y saquinavir (63%) después de su combinación con enfuvirtida. Se desconoce el mecanismo de esta interacción. No se recomienda el ajuste de dosis de tipranavir o ritonavir. Nucleósidos inhibidores de la transcriptasa reversa: zidovudina (ZDV): APTIVUS®, coadministrado con ritonavir a dosis bajas, disminuye el ABC de zidovudina aproximadamente un 35%. Esto no impacta los niveles de ZDV glucuronizado. La relevancia clínica en la reducción en los niveles plasmáticos de zidovudina no ha sido establecida y un ajuste en la dosis de zidovudina no puede ser recomendado en este momento. Didanosina: APTIVUS®, coadministrado con ritonavir a dosis bajas, provoca una reducción en el ABC de la didanosina. La importancia clínica de la reducción de didanosina en los niveles de plasma aún no se ha establecido. La dosificación de la didanosina con recubrimiento entérico y APTIVUS®, coadministrado con ritonavir a dosis bajas, debe administrarse por separado al menos con 2 horas de diferencia para evitar incompatibilidad de fórmulas. Lamivudina y estavudina: APTIVUS®, coadministrado con ritonavir a dosis bajas, no provoca un cambio significativo en el ABC de lamivudina o estavudina, por lo que no se recomienda un ajuste de la dosis de lamivudina o estavudina. Abacavir: APTIVUS®, coadministrado con ritonavir a dosis bajas, disminuye aproximadamente en un 40% el ABC de abacavir. La importancia clínica en la reducción en los niveles de abacavir no se ha establecido y no se recomienda un ajuste de dosis de abacavir en este momento. Inhibidores nucleótidos de la trascriptasa inversa: tenofovir: APTIVUS®, coadministrado con ritonavir a dosis bajas, no provocó un cambio significativo en las concentraciones en plasma de tenofovir; no se recomienda un ajuste de la dosis de tenofovir. Inhibidores no nucleótidos de la trascriptasa inversa: nevirapina: no se ha observado interacción significativa entre APTIVUS®, coadministrado con ritonavir a dosis bajas, y nevirapina. Por lo tanto, no es necesario realizar ajustes en la dosificación. Efavirenz: el estado estable de 600 mg de efavirenz administrado diariamente coadministrado con APTIVUS® y ritonavir a dosis bajas (500 mg/200mg cada 12 horas) no tiene un impacto significativo sobre el ABC y la Cmáx del tipranavir (disminuciones del 2,9% y 8,3%, respectivamente) y resulta en un aumento clínicamente insignificante en la Cp12h (19,2%). APTIVUS® coadministrado con ritonavir a dosis bajas no tiene un impacto significativo sobre el ABC y la Cmín de efavirenz. Inhibidores de la proteasa: amprenavir, atazanavir, lopinavir, saquinavir: en un estudio clínico de terapia combinada con un inhibidor de la proteasa de doble esfuerzo en tratamiento múltiple realizado en adultos VIH positivos, APTIVUS® coadministrado con ritonavir a dosis bajas, provocó una reducción del 55%, 70% y 78% en la Cmín de amprenavir, lopinavir y saquinavir, respectivamente. Fue observada similarmente una reducción del 81%. en la Cmín de atazanavir en un estudio realizado a voluntarios sanos. Por lo tanto, no se recomienda la administración concomitante de APTIVUS® coadministrado con ritonavir a dosis bajas con amprenavir/ritonavir, atazanavir/ritonavir, lopinavir/ritonavir o saquinavir/ritonavir; la importancia clínica de la reducción en sus niveles aún no se ha establecido. Si se considera absolutamente necesario la administración concomitante, no puede recomendarse un ajuste de la dosis. Actualmente, no se encuentra disponible ningún dato sobre las interacciones de APTIVUS®, coadministrado con ritonavir a dosis bajas, dosificado concomitantemente con inhibidores de proteasa diferentes a aquellos anteriormente mencionados. Anticonvulsivantes: la carbamazepina, el fenobarbital y la fenitoína deben usarse con cuidado en combinación con APTIVUS®/ritonavir. El uso concomitante de carbamazepina a una dosis de 200 mg dos veces al día resultó en aumento en las concentraciones plasmáticas de la carbamazepina (en aproximadamente 23% en la media geométrica de la Cmín para el total de carbamazepina y el carbamazepina-10,11-epóxido; ambos son mitades activas farmacológicamente) y una disminución en la Cmín del tipranavir (en aproximadamente el 61% comparada con los controles históricos), lo cual podría resultar en disminución de su efectividad. Antifúngicos: fluconazol: APTIVUS®, coadministrado con ritonavir a dosis bajas, no afecta substancialmente la farmacocinética en estado estable del fluconazol. El fluconazol incrementa el ABC y la Cmín de APTIVUS® en un 56% y en 104%, respectivamente, cuando se comparó con los antecedentes. No se recomiendan un ajuste en la dosis. Tampoco se recomiendan la administración de dosis de fluconazol > 200 mg/día. Itraconazol y ketoconazol: con base a las concentraciones teóricas del APTIVUS® coadministrado con ritonavir a dosis bajas, se espera que aumenten las concentraciones de itraconazol y ketoconazol. El itraconazol y ketoconazol debe utilizarse con precaución (no se recomiendan la administración de dosis de ketoconazol > 200 mg/día). Voriconazol: debido a que hay involucrados múltiples sistemas de isoenzimas del CYP en el metabolismo de voriconazol, es difícil predecir la interacción con el APTIVUS® coadministrado con ritonavir a dosis bajas. Inhibidores de HMG CoA reductasa: atorvastatina: APTIVUS®, coadministrado con ritonavir a dosis bajas, incrementa las concentraciones plasmáticas de atorvastatina aproximadamente entre 8-10 veces y reduce las ABCs de los metabolitos hidroxilo en aproximadamente un 85%. La atorvastatina no modifica significativamente el ABC, la Cmáx o la Cmín de APTIVUS®. Se recomienda iniciar el tratamiento con la dosis mínima posible de atorvastatina junto con un monitoreo cuidadoso o considerar otros inhibidores de HMG-CoA reductasa como pravastatina, fluvastatina o rosuvastatina. Inductores de la isoenzima CYP: rifabutina: la concentración de rifabutina en el plasma se incrementa hasta tres veces por la administración de APTIVUS® coadministrado con ritonavir a dosis bajas y hasta 20 veces la del metabolito activo 25-Odesacetil-rifabutina. La rifabutina incrementa en un 16% la Cmín de APTIVUS®. Se recomiendan una reducción en la dosis de rifabutina al menos de un 75% de los 300 mg/día, usualmente utilizados (es decir, 150 mg en días alternos o tres veces a la semana). Los pacientes que reciben rifabutina con APTIVUS®/ritonavir deberán ser monitoreados de cerca por un surgimiento de evento adverso con la terapia de rifabutina. Puede ser necesaria una reducción adicional de la dosis anteriormente mencionada. Inhibidores de isoenzima CYP: claritromicina: APTIVUS®, coadministrado con ritonavir a dosis bajas, incrementa el ABC y la Cmín de la claritromicina en un 19% y 68%, respectivamente, y disminuye el ABC del metabolito activo 14-hidroxi alrededor de un 95%. Estos cambios no se consideran clínicamente importantes. La claritromicina aumenta la Cmín de APTIVUS® en más del 100%. Este incremento importante en la Cmín puede ser clínicamente relevante. Los pacientes que utilizan la claritromicina en dosis mayores a 500 mg dos veces al día deben ser cuidadosamente monitorizados para vigilar si se presenta cualquier signo de toxicidad. En el caso de los pacientes con insuficiencia renal, deben considerarse los siguientes ajustes en la dosis: para los pacientes con una depuración de creatinina (CLCR, por sus siglas en inglés) de 30 a 60 ml/min., la dosis de claritromicina debe reducirse en un 50%. Para los pacientes con una depuración de creatinina (CLCR, por sus siglas en inglés) < 30 ml/min., la dosis de claritromicina debe reducirse en un 75%. No son necesarios los ajustes de dosis para los pacientes con función renal normal. Anticonceptivos orales/estrógenos: APTIVUS®, coadministrado con ritonavir a dosis bajas, disminuye el ABC y la Cmáx del etinil-estradiol en un 50%, pero no altera significativamente el comportamiento farmacocinética de la noretindrona. Deben utilizarse medidas anticonceptivas alternas o adicionales cuando los anticonceptivos orales basados en estrógenos se administra con APTIVUS® coadministrado con ritonavir a dosis bajas. Los pacientes que usan estrógenos como terapia hormonal de reemplazo deberán ser clínicamente monitoreados por señales de deficiencia de estrógenos. Debe hacerse notar que otros compuestos que son substratos de CYP3A pueden incrementar las concentraciones en plasma cuando se administra con APTIVUS® coadministrado con ritonavir a dosis bajas. Inhibidores de la bomba de protones: omeprazol: APTIVUS® coadministrado con ritonavir a dosis bajas disminuye el ABC y la Cmáx del omeprazol en 71% y 73%, respectivamente. No se observaron cambios clínicamente importantes en el estado estable del tipranavir/ritonavir. La dosis de omeprazol puede necesitar incrementarse cuando se coadministra con APTIVUS® y ritonavir. Otros agentes: midazolam: el uso concomitante de APTIVUS®/ritonavir y el midazolam oral está contraindicado. El ritonavir es un potente inhibidor del CYP3A, y por lo tanto, afectará a los medicamentos metabolizados por esta enzima. Las concentraciones de una sola dosis de midazolam administrada vía intravenosa aumentaron 2,8 veces (ABC 0-24h) cuando se coadministró con APTIVUS®/ritonavir en estado estable. Si el APTIVUS®/ritonavir se coadministra con midazolam parenteral, debe instituirse vigilancia clínica estrecha por depresión respiratoria y/o sedación prolongada y realizar los ajustes de dosis pertinentes. Inmunosupresores (ciclosporina, tacrolimus, sirolimus): el efecto de la coadministración de APTIVUS®, coadministrado con ritonavir a dosis bajas sobre un substrato para el CYP3A4/5, muestra potente inhibición en la primera dosis y en estado estable del APTIVUS®/ritonavir. Cuando APTIVUS®/ritonavir se coadministra con un substrato para P-gp, ocurre inhibición moderada del P-gp con la primera dosis de APTIVUS®/ritonavir; sin embargo, no hay efecto sobre la P-gp con el estado estable del APTIVUS®/ritonavir. Se anticipa que se verán efectos similares con estos inmunosupresores. Se recomienda la vigilancia de las concentraciones de estos inmunosupresores cuando estos productos medicinales se combinen con APTIVUS®/ritonavir. Warfarina y otros anticoagulantes orales: el efecto de APTIVUS®, coadministrado con ritonavir a dosis bajas sobre la S-warfarina, resultó en un incremento del 18% en la exposición de la S-warfarina con la primera dosis de APTIVUS®/ritonavir, y un disminución del 12% en la exposición de la S-warfarina con el estado estable del APTIVUS®/ritonavir. Se recomienda un monitoreo cercano clínico y biológico (medición INR) cuando se combinan estos productos medicinales. Teofilina: se espera que APTIVUS® coadministrado con ritonavir a dosis bajas disminuya las concentraciones de teofilina. Pueden requerirse un incremento en la dosis de teofilina y debe considerarse el monitoreo terapéutico. Desipramina: se recomienda la reducción de la dosis y el monitoreo de la concentración de desipramina, debido a que se espera que APTIVUS®, coadministrado con ritonavir a dosis bajas, incremente las concentraciones de desipramina. Loperamida: un estudio de interacción farmacodinámica realizado en voluntarios sanos demostró que la administración de loperamida y APTIVUS®, coadministrado con ritonavir a dosis bajas, no provoca cambios clínicamente relevantes en la respuesta respiratoria al dióxido de carbono. El análisis farmacocinética mostró que el ABC y la Cmáx de loperamida se redujeron en un 51% y 61%, respectivamente, y en tanto que la Cmín de APTIVUS® en un 26%. Se desconoce la importancia clínica de estos hallazgos. Tadalafil: el uso concomitante de APTIVUS®, coadministrado con ritonavir a dosis bajas con tadalafil, resultó en un incremento de 2,3 veces en la exposición del tadalafil con la primera dosis de APTIVUS®/ritonavir y sin cambios en la exposición del tadalafil con el estado estable del tipranavir/ritonavir. Si se usa tadalafil dentro de los primeros días del tratamiento con APTIVUS®/ritonavir, entonces debe administrarse la menor dosis. Sin embargo, después de 7 a 10 días de administrar APTIVUS®/ritonavir, y cuando se alcance el estado estable para tipranavir y ritonavir, la dosis de tadalafil puede aumentarse, si se requiere.
Alteraciones en los resultados de pruebas de laboratorio: Las frecuencias de las alteraciones en los resultados de las pruebas de laboratorio clínico (Grado 3 o 4) reportadas por lo menos en 2% de los pacientes en los brazos del APTIVUS®, coadministrado con ritonavir a dosis bajas en los estudios clínicos fase III (RESIST-1 y RESIST-2), fueron: AST incrementada (6,1%), ALT incrementada (9,6%), amilasa incrementada (6,0%), colesterol incrementado (4,2%), triglicéridos aumentados (24,9%) y disminución en los conteos leucocitarios (5,7%). En los estudios clínicos RESIST-1 y RESIST-2 extendidos hasta las 96 semanas, la proporción de pacientes quienes desarrollaron elevaciones grados 2 a 4 en la ALT y/o la AST aumentaron de 26% en la semana 48 a 29,3% en la semana 96 con el APTIVUS®/ritonavir y de 13,7% en la semana 48 a 14,6% en la semana 96 con el IP/ritonavir comparador, demostrando que el riesgo de desarrollar elevaciones en las transaminasas durante el segundo año de terapia es menor que durante el primer año. Las elevaciones grados 3/4 de ALT y/o AST con APTIVUS coadministrado con ritonavir a bajas dosis continúo aumentando de 10% en la semana 48 a 14.7% en la semana 96, y para el IP/ritonavir comparador de 3,4% a 4,5% en las semanas 48 y 96, respectivamente.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Se han realizado estudios de toxicología en animales con APTIVUS® solo y coadministrado con ritonavir (3,75:1 relación peso/peso) en varias especies. Los estudios con la coadministración de APTIVUS® y ritonavir no revelaron efectos toxicológicos adicionales cuando se compararon con aquellos observados en los estudios toxicológicos de APTIVUS® solo. Los efectos predominantes de la administración repetida de APTIVUS® en todas las especies analizadas toxicológicamente fueron sobre el tracto gastrointestinal (emesis, evacuaciones sueltas, diarrea) y el hígado (hipertrofia). Cuando terminó el tratamiento, los efectos fueron reversibles. Cambios adicionales incluyeron hemorragia en ratas a altas dosis (específicamente roedores). Se observó hemorragia en ratas asociada con tiempo de protrombina (TP) prolongado y activación parcial del tiempo de tromboplastina (APTT). La mayoría de los efectos en estudios de toxicidad en dosis repetidas aparecieron en los niveles de exposición sistémica que son equivalentes o por debajo de los niveles de exposición en humanos se recomienda una dosis clínica. No se observaron eventos adversos en el apareamiento o en la fertilidad en un estudio realizado en ratas con APTIVUS® a niveles de exposición sistémica (ABC) equivalentes a la exposición en humanos con la dosis clínica en humano adulto. El APTIVUS® no produjo efectos teratogénicos a las dosis maternas que producen niveles de exposición sistémica del fármaco aproximadamente equivalentes o debajo de la dosis clínica utilizada en humano adulto (APTIVUS® coadministrado con ritonavir a dosis bajas 500 mg/200 mg administrados dos veces al día). Al exponer a la dosis clínica de APTIVUS® a ratas aproximadamente 0,8 veces la exposición en humanos, se observó toxicidad fetal (decremento en osificación y peso en el cuerpo). En los estudios de desarrollo pre y postnatal con APTIVUS® en ratas, no se observaron efectos adversos en la exposición de 0,2 veces la exposición en humanos, pero se observó la inhibición en el crecimiento de las crías a la dosis maternalmente tóxicas cercanas a 0,8 veces la exposición humana. El bioensayo de la carcinogenicidad en un largo plazo en ratas y en ratones se ha completado y reveló potencial específico tumorigénico para estas especies, que no se considera de relevancia clínica. APTIVUS® no mostró evidencia de toxicidad genética en una batería de cinco pruebas in vitro y en pruebas in vivo que evaluaron la mutagenicidad y la clastogenicidad. En estudios preclínicos, el tratamiento con APTIVUS® indujo cambios en los parámetros de coagulación (incremento en el tiempo de protrombina y tiempo parcial de tromboplastina) en roedores. A grandes dosis y en casos extremos, estos cambios produjeron sangrado en múltiples órganos y la muerte. El mecanismo de producción de estos efectos es desconocido. No se ha observado algún efecto sobre los parámetros de coagulación en estudios preclínicos llevados a cabo en perros con tipranavir. La coadministración de tipranavir y vitamina E no se ha estudiado en perros. La evaluación clínica de los efectos sobre la coagulación en pacientes infectados con el VIH-1 no demostró un efecto del tipranavir más ritonavir.
Dosis y vía de administración: Adultos: la dosis recomendada de APTIVUS® es de 500 mg (dos cápsulas de 250 mg), coadministrado con 200 mg de ritonavir (ritonavir en baja dosis), dos veces al día. APTIVUS® debe administrarse con dosis bajas de ritonavir para asegurar su efecto terapéutico. Debe instruirse adecuadamente a los pacientes sobre la forma correcta de dosificación. Consulte también la información para prescribir de ritonavir con el objetivo de revisar las contraindicaciones, precauciones generales, reacciones adversas e interacciones potenciales del medicamento. APTIVUS® debe ser coadministrado con ritonavir a dosis bajas y deben administrarse junto con los alimentos para mejorar la tolerancia al ritonavir. Terapia concomitante: APTIVUS®, coadministrado con ritonavir a dosis bajas, deberá administrarse por lo menos con dos agentes antirretrovirales adicionales. Debe consultarse la información para prescribir de los agentes antirretrovirales utilizados.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: No existe un antídoto conocido para la sobredosis con APTIVUS®. El tratamiento deberá consistir en medidas generales de soporte.
Presentación(es): Caja con frasco con 120 cápsulas.
Recomendaciones sobre almacenamiento: APTIVUS® debe conservarse en un refrigerador de 2°C a 8°C. Consérvese el frasco bien tapado.
Leyendas de protección: Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. No se use en el embarazo ni en la lactancia.
Nombre y domicilio del laboratorio: Hecho en Estados Unidos de América por: Cardinal Health PTS, LLC 2725 Scherer Drive St. Petersburg, Florida 33716-1016. Acondicionado en Estados Unidos de América por: 1809 Wilson Road Columbus, Ohio 43228 Roxane Laboratories Inc. Distribuido por: Boehringer Ingelheim Promeco, S.A. de C.V., Maíz No. 49, Barrio Xaltocán, 16090, México, D.F. ®Marca registrada.
Número de registro del medicamento: 232M2005 SSA IV.
Clave de IPPA: FEAR-0533002050089/R2005
APTIVUS*
BOEHRINGER PM
Solución
Denominación genérica: Tipranavir.
Forma farmacéutica y formulación: Solución. Cada 100ml contienen: tipranavir 10 g. Vehículo cbp 100 ml.
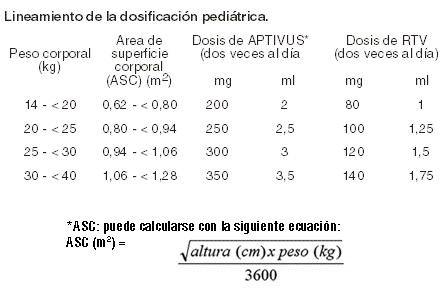
Indicaciones terapéuticas: APTIVUS* está indicado para el tratamiento de pacientes infectados con VIH-1 mayores de 6 años de edad que han estado bajo tratamiento con un inhibidor de proteasa. APTIVUS* debe utilizarse en combinación con una dosis de 200 mg de ritonavir (RTV) dos veces al día. Al decidir sobre un nuevo régimen de tratamiento para los pacientes que no han estado bajo tratamiento con un régimen antirretroviral, debe realizarse una cuidadosa valoración individual del historial de tratamiento del paciente y de los modelos de mutaciones asociados con los diferentes fármacos. Puede ser apropiado utilizar las pruebas de resistencia cuando éstas se encuentren disponibles.
Farmacocinética y farmacodinamia: Mecanismo de acción: el virus de la inmunodeficiencia humana (VIH-1) codifica una proteasa aspartilo que es esencial para el corte y la maduración de los precursores de la proteína viral. El tipranavir, principio activo de APTIVUS*, es un inhibidor no peptídico de la proteasa del VIH-1 que inhibe la replicación viral evitando la maduración de las partículas virales. Actividad antiviral in vitro: el tipranavir inhibe la replicación de las cepas del laboratorio del VIH-1 y los aislados clínicos en modelos agudos de infección de células T, con concentraciones efectivas en un 50% (CE50) que varían de 0,03 a 0,07 mM (18-42 ng/ml). El tipranavir también es efectivo en la inhibición de la replicación de las cepas M-trópicas de VIH (CE90 ADA = 0,75 mM, 452 ng/ml y CE90 DGV = 0,3 mM, 180 ng/ml) y en la inhibición de la acumulación extracelular de la proteína p24 de la cápsida de las células H-9 infectadas de forma crónica por el VIH-1 IIIB (CE50 de 0,39 mM, 235 ng/ml). Los estudios de unión a proteínas han demostrado que la actividad antiviral del tipranavir disminuye en un promedio de 3,75 veces en condiciones donde se encuentra presente el suero humano. Cuando se utiliza en combinación con otros antirretrovirales, el tipranavir muestra una sinergia a la adición con un inhibidor nucleósido de transcriptasa reversa (INTR) zidovudina, un inhibidor no nucleósido de transcriptasa reversa (INNTR) delavirdina y el inhibidor de la proteasa, ritonavir. Se reportaron actividades que variaron de la sinergia al antagonismo cuando el tipranavir se utilizó en combinación con los inhibidores de la proteasa, lopinavir y amprenavir. Resistencia: el desarrollo de la resistencia in vitro a tipranavir es lento y complejo. En un experimento particular de resistencia in vitro, se seleccionó un aislado del VIH-1 que fue 87 veces más resistente al tipranavir después de 9 meses, y contenía 10 mutaciones en la proteasa: L10F, I13V, V32I, L33F, M36I, K45I, I54V/T, A71V, V82L, I84V así como una mutación en el sitio de corte CA/P2 de la poliproteína gag. Experimentos genéticos inversos demostraron que fue necesaria la presencia de 6 mutaciones en la proteasa (I13V, V32I, L33F, K45I, V82L, I84V) para conferir > 10 veces la resistencia al tipranavir mientras que el genotipo de mutación 10 confirió una resistencia de 69 veces a tipranavir. Existe una correlación inversa in vitro, entre el grado de resistencia al tipranavir y la capacidad de los virus para replicarse. Los virus recombinantes que muestran una resistencia ≥3 veces a tipranavir crecen menos del 1% de la relación detectada para el VIH-1 tipo salvaje en las mismas condiciones. Los virus resistentes al tipranavir que surgen in vitro del VIH-1 tipo salvaje muestran una disminución en la susceptibilidad a los inhibidores de la proteasa, amprenavir, atazanavir, indinavir, lopinavir, nelfinavir y ritonavir pero permanecen sensibles a saquinavir. In vivo, se observó también el surgimiento de mutaciones en las posiciones 33, 82 y 84 de la proteasa después del tratamiento con tipranavir. A través de una serie de múltiples análisis de regresión escalonada de genotipos al inicio del tratamiento y durante el mismo a partir de todos los estudios clínicos, se han asociado 16 aminoácidos con una reducción a la susceptibilidad al tipranavir y/o reducción de la respuesta de la carga viral a las 24 semanas: 10V, 13V, 20M/R/V, 33F, 35G, 36I, 43T, 46L, 47V, 54A/M/V, 58E, 69K, 74P, 82L/T, 83D y 84V. Los aislados clínicos que mostraron una disminución en la susceptibilidad al tipranavir incluyeron más de siete mutaciones asociadas con tipranavir. En los estudios clínicos Fase II, 217 pacientes con genotipos bajo tratamiento han demostrado que las mutaciones predominantes que surgen con el tratamiento con APTIVUS* son L33F/I, V82T/L y I84V. Se requiere generalmente la combinación de estos tres tipos para una susceptibilidad reducida. Las mutaciones en los codones L33F/I y I84V se encuentran entre las primeras que se presentan. Las mutaciones en la posición 82 se presentan por medio de dos rutas: una a partir de la mutación preexistente de 82A seleccionando a 82T, la otra del tipo salvaje 82V seleccionando a 82L. Resistencia cruzada: el tipranavir mantiene una importante actividad antiviral (resistencia < 4 veces) contra la mayoría de los aisla