ARIMIDEX®
ASTRAZENECA
Denominación genérica: Anastrozol.
Forma farmacéutica y formulación: Tabletas. Cada tableta contiene: Anastrozol 1 mg. Excipiente cbp Una tableta.
Indicaciones terapéuticas: Tratamiento de cáncer de mama temprano en mujeres postmenopáusicas. En pacientes con receptores hormonales positivos se han observado los beneficios durante el tratamiento con ARIMIDEX®. Reducción en la incidencia de cánceres de mama contralaterales en pacientes que reciben ARIMIDEX® como tratamiento adyuvante de cáncer de mama temprano.
Tratamiento de cáncer de mama avanzado en mujeres postmenopáusicas.
Farmacocinética y farmacodinamia: Farmacocinética: Después de su administración oral, la absorción de anastrozol es rápida y las concentraciones plasmáticas máximas ocurren típicamente dentro de las primeras dos horas (en condiciones de ayuno). Anastrozol se elimina lentamente con una vida media de eliminación plasmática de 40-50 horas. La ingestión de alimento disminuye ligeramente la velocidad pero no el grado de absorción. No se espera que el cambio menor en la tasa de absorción tenga un efecto clínicamente significativo sobre la concentración plasmática del estado estable para una dosis diaria de tabletas de ARIMIDEX®. Después de siete días se obtiene el 90-95% de la concentración plasmática del estado estable de anastrozol en plasma. No existe evidencia de la dependencia en función del tiempo o de la dosis con base en parámetros farmacocinéticos de anastrozol. La farmacocinética de anastrozol es independiente de la edad en mujeres postmenopáusicas. En niños con ginecomastia en la pubertad, anastrazol fue rápidamente absorbido, ampliamente distribuido y eliminado lentamente, con una vida media de aproximadamente 2 días. Los parámetros farmacocinéticos en niños fueron comparables con aquéllos de mujeres postmenopáusicas. La depuración de anastrazol fue menor en niñas que en niños y la exposición fue más elevada. Anastrazol en niñas se distribuyó más ampliamente y se eliminó lentamente con una vida media estimada de aproximadamente 0.8 días. La unión a proteínas plasmáticas de anastrozol es de 40%. Anastrozol se metaboliza ampliamente en mujeres postmenopáusicas donde menos del 10% del compuesto original sin cambio se elimina por la orina dentro de las 72 horas siguientes a su administración. El metabolismo de anastrozol se realiza por N-desalquilación, hidroxilación y glucuronización. Los metabolitos son excretados principalmente por la orina. Triazol, el metabolito principal en plasma y orina, no inhibe a la aromatasa. Después de su administración oral, la absorción de anastrozol en voluntarios con cirrosis hepática estable o insuficiencia renal se mantuvo dentro del rango observado en voluntarios sanos. Farmacodinamia: ARIMIDEX® es un potente y altamente selectivo inhibidor de aromatasa no esteroideo. En mujeres postmenopáusicas la producción de estradiol resulta de la conversión de androstenediona a estrona a través del complejo enzimático de la aromatasa en los tejidos periféricos. Estrona es subsecuentemente convertida en estradiol. La reducción de los niveles circulantes de estradiol ha demostrado que produce un efecto benéfico en mujeres con cáncer de mama. En mujeres postmenopáusicas, ARIMIDEX® en una dosis diaria de 1 mg produjo una supresión de estradiol mayor al 80% utilizando una prueba altamente sensible. ARIMIDEX® no posee actividad progestogénica, androgénica o estrogénica. Dosis diarias de hasta 10 mg de ARIMIDEX® no tienen ningún efecto sobre la secreción de cortisol o aldosterona con base en determinaciones anteriores y posteriores con pruebas de reto con ACTH. Por lo tanto, no se requiere suplemento corticoide. Un amplio programa de estudios clínicos fase III mostró que ARIMIDEX® es un tratamiento efectivo para el cáncer de mama temprano y el cáncer de mama avanzado en mujeres postmenopáusicas susceptibles de tratamiento endocrino. Tratamiento adyuvante primario en cáncer de mama: En un amplio estudio fase III realizado en 9,366 mujeres postmenopáusicas con cáncer de mama operable y en tratamiento durante 5 años, ARIMIDEX® mostró ser estadísticamente superior a tamoxifeno en lo que respecta a periodo de supervivencia libre de enfermedad. Para la población definida prospectivamente como positiva a receptores hormonales, se observó un mayor beneficio en lo que se refiere al periodo de supervivencia libre de enfermedad a favor de ARIMIDEX® en comparación con tamoxifeno. Estadísticamente, ARIMIDEX® fue superior a tamoxifeno en tiempo para la recurrencia. La diferencia fue de mayor magnitud que en cuanto a periodo libre de enfermedad, tanto en la población con Intención de Tratamiento (ITT) como en la población con receptores hormonales positivos. ARIMIDEX® fue estadísticamente superior a tamoxifeno en términos de tiempo para la recurrencia a distancia. Existe también una tendencia numérica a favor de la supervivencia libre de enfermedad a distancia. La incidencia de cáncer de mama contralateral tuvo una reducción estadísticamente significativa con ARIMIDEX® en comparación con tamoxifeno. El beneficio de supervivencia global con tamoxifeno se mantuvo con ARIMIDEX®. Un análisis adicional de tiempo hasta la muerte después de la recurrencia, mostró una tendencia numérica a favor de ARIMIDEX® en comparación con tamoxifeno. ARIMIDEX® es bien tolerado. Se han reportado los siguientes eventos adversos a pesar de la causalidad. Las pacientes que reciben ARIMIDEX® presentan reducción de bochornos, sangrado vaginal, desecho vaginal, cáncer endometrial, eventos tromboembólicos venosos y cerebrovasculares en comparación con las pacientes que reciben tamoxifeno. Las pacientes que reciben ARIMIDEX® presentan un incremento en afecciones en las articulaciones (incluyendo artritis, artrosis y artralgia) y fracturas comparadas con pacientes que reciben tamoxifeno. Se observó una proporción de fracturas de 22 por 1000 años-paciente con ARIMIDEX® y 15 por 1000 años-paciente con el grupo de tamoxifeno con una mediana de seguimiento de 68 meses. La proporción de fracturas con ARIMIDEX® cae dentro del rango amplio de la proporción de fracturas reportadas a una edad que coincide con la población postmenopáusica. La combinación de ARIMIDEX® y tamoxifeno, en todas las pacientes así como en las pacientes con receptores hormonales positivos, no demostró ningún beneficio en la eficacia en comparación con tamoxifeno. Este brazo del tratamiento fue discontinuado del estudio. Tratamiento adyuvante de cáncer de mama temprano para pacientes que están siendo tratadas con tamoxifeno como adyuvante: En un estudio clínico fase III (ABCSG 8) conducido en 2579 mujeres postmenopáusicas con cáncer de mama temprano con receptores hormonales positivos que están siendo tratadas con tamoxifeno como adyuvante, las pacientes tuvieron una supervivencia libre de enfermedad superior cuando se cambiaron a ARIMIDEX® en comparación con aquéllas que continuaron con tamoxifeno. El tiempo hasta cualquier recurrencia, el tiempo a recurrencia local o distante y el tiempo a la recurrencia distante confirmaron una ventaja estadística para ARIMIDEX®, consistente con los resultados de supervivencia libre de enfermedad. La incidencia de cáncer contralateral fue muy baja en los dos brazos del tratamiento con una ventaja numérica para ARIMIDEX®. La supervivencia total fue similar para los dos grupos en tratamiento. Dos estudios clínicos similares con ARIMIDEX® (GABG/ARNO 95 e ITA), así como el análisis combinado de ABCSG 8 y GABG/ARNO 95 apoyaron estos resultados. El perfil de seguridad de ARIMIDEX® en estos tres estudios fue consistente con el perfil de seguridad conocido establecido en mujeres postmenopáusicas con cáncer de mama temprano con receptores hormonales positivos. Estudio de anastrazol con bifosfonato risedronato (SABRE): Densidad Mineral Osea (DMO). En el estudio SABRE fase III/IV, 243 mujeres postmenopáusicas con cáncer de mama temprano con receptores hormonales positivos fueron estratificadas para tratamiento con ARIMIDEX® en grupos de bajo, moderado y alto riesgo de acuerdo con el riesgo existente de fractura por fragilidad. Todas las pacientes recibieron tratamiento con vitamina D y calcio. Las pacientes del grupo de bajo riesgo recibieron solamente ARIMIDEX®, las del grupo moderado fueron aleatorizadas a ARIMIDEX® mas bifosfonato o ARIMIDEX® mas placebo y aquéllas del grupo de alto riesgo recibieron ARIMIDEX® más bifosfonato. El análisis principal a 12 meses mostró que las pacientes que ya tenían riesgo de moderado a alto de fractura por fragilidad (evaluada mediante densidad mineral del hueso y formación del hueso y marcadores de reabsorción), habían manejado la salud de sus huesos exitosamente, empleando ARIMIDEX® en combinación con bifosfonato. Adicionalmente, ningún cambio en DMO fue visto en el grupo de bajo riesgo tratado sólo con ARIMIDEX®, vitamina D y calcio. Estos hallazgos fueron reflejados en la variable de eficacia secundaria del cambio de la línea basal en la DMO total de la cadera a los 12 meses. Este estudio proporciona evidencia de que las mujeres postmenospáusicas con cáncer de mama temprano, programadas para ser tratadas con ARIMIDEX®, deberán manejar el estado de sus huesos de acuerdo con los lineamientos disponibles para mujeres postmenopáusicas en riesgo similar de fractura por fragilidad. Lípidos: En el estudio SABRE hubo un efecto neutro en lípidos plasmáticos tanto en aquellas pacientes tratadas únicamente con ARIMIDEX® como en aquéllas tratadas con ARIMIDEX® más un bifosfanato. Pediátricos: Se realizaron 3 estudios clínicos en pacientes pediátricos (2 en niños en pubertad con ginecomastia y 1 pediátrico en niñas con el síndrome McCune Albright). Estudio de Ginecomastia: El estudio 0006 fue un estudio multi-centro, aleatorizado, doble-ciego, en 80 niños púber con ginecomastia de más de 12 meses de duración (edades inclusive de 11 a 18 años) tratados con ARIMIDEX® 1 mg/día o placebo, diariamente hasta por 6 meses. Se observó una reducción > 50% en el volumen total de la mama, medido mediante ultrasonido, en el 38.5% (15/39) del grupo de ARIMIDEX® y 31.4% (11/35) en el grupo tratado con placebo (proporción = 1.513, IC 95% 0.496 a 4.844, p= 0.4687). El estudio 0001 fue un estudio abierto, farmacocinético (PK) de dosis-múltiple con ARIMIDEX® 1 mg/día en 36 niños púberes con ginecomastia de menos de 12 meses de duración. Una reducción del 50% o más en el volumen total de la mama a los 6 meses, se observó en el 55.6% (20/36) de los niños. Estudio del Síndrome McCune Albright (SMA): El estudio 0046 fue un estudio exploratorio, abierto, multi-centro e internacional de ARIMIDEX® en 28 niñas (edad 2 a ≤ 10 años) con Síndrome McCune-Albright (SMA). No se observaron cambios estadísticamente significativos en la frecuencia de los días de sangrado vaginal durante el tratamiento. De las pacientes con sangrado vaginal en la línea base, 28% experimentaron una reducción ≥ 50% en la frecuencia de días de sangrado en el tratamiento, 40% experimentaron una cesación sobre un periodo de 6 meses y 12% experimentaron cesación sobre un periodo de 12 meses. No hubo cambios clínicamente significativos en la clasificación Tanner, volumen ovárico medio o volumen uterino medio. No se observó ningún cambio estadísticamente significativo en la velocidad de incremento de la edad del hueso durante el tratamiento, en comparación con la velocidad de la línea base. La velocidad de crecimiento (en cm/año) se redujo significativamente (p < 0.05) a partir del pre-tratamiento de 0 a 12 meses y a partir del pre-tratamiento al segundo semestre (mes 7 a mes 12). La evaluación total de los EAs en niños de menos de 18 años de edad, no generó preocupaciones de seguridad y tolerabilidad.
Contraindicaciones: ARIMIDEX® no debe ser administrado durante el embarazo o la lactancia. Hipersensibilidad conocida a la sustancia activa o a cualquiera de los componentes del producto.
Precauciones generales: ARIMIDEX® no se recomienda para el uso en niños o mujeres premenopáusicas ya que la seguridad y eficacia no han sido establecidas en este grupo de pacientes. (Véase Farmacocinética y farmacodinamia). ARIMIDEX® no ha sido investigado en pacientes con insuficiencia hepática o renal severa. El potencial riesgo/beneficio para tales pacientes debe ser cuidadosamente considerado antes de la administración de ARIMIDEX®. Como ARIMIDEX® disminuye los niveles de estrógenos circulantes, puede ocasionar una reducción en la densidad mineral ósea teniendo como probable consecuencia un incremento en el riesgo de fracturas. Este posible incremento de riesgo debe manejarse de acuerdo a las guías de tratamiento para la salud ósea en mujeres posmenopausicas. Efectos en la habilidad para conducir u operar maquinaria: Es poco factible que ARIMIDEX® altere la capacidad para conducir un vehículo u operar maquinaria. Sin embargo, astenia y somnolencia han sido asociadas al uso de ARIMIDEX®, por lo que se debe tener precaución al conducir un vehículo u operar maquinaria mientras tales síntomas persistan.
Restricciones de uso durante el embarazo y la lactancia: ARIMIDEX® no debe ser administrado durante el embarazo o la lactancia.
Reacciones secundarias y adversas: A menos que se especifique, las siguientes categorías de frecuencia fueron calculadas a partir del número de eventos adversos reportados en un estudio fase III realizado en 9366 mujeres postmenopáusicas con cáncer de mama operable tratado durante 5 años y únicamente si se indica, bajo ningún concepto fue tomada de la frecuencia del grupo de tratamiento comparativo o si el investigador consideraba que estaba relacionado a la medicación en estudio.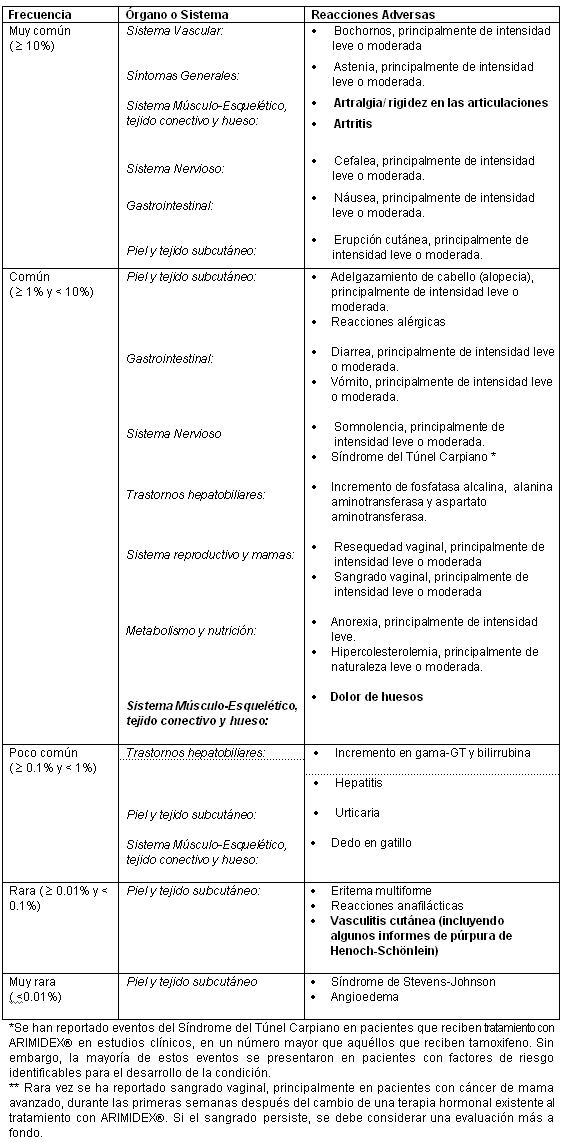
En un estudio Fase III conducido en 9366 mujeres postmenopáusicas con cáncer de mama operable, en tratamiento durante 5 años, se reportaron eventos cardiovasculares isquémicos con más frecuencia en pacientes tratados con ARIMIDEX® en comparación con aquéllos tratados con tamoxifeno, aunque la diferencia no fue estadísticamente significativa. La diferencia observada fue principalmente debida a más reportes de angina de pecho y estuvo asociada con un subgrupo de pacientes con enfermedad isquémica cardíaca pre-existente.
Interacciones medicamentosas y de otro género: Los estudios clínicos de interacción con antipirina y cimetidina indican que es poco probable que la coadministración de ARIMIDEX® con otros medicamentos produzca interacciones medicamentosas mediadas por el citocromo P450 que sean clínicamente significativas. La revisión de los aspectos de seguridad en la base de datos clínicos no reveló evidencia de interacciones clínicas significativas en pacientes tratadas con ARIMIDEX® quienes también recibieron otros medicamentos comúnmente prescritos. No hubo interacciones clínicamente significativas con bifosfonatos (Véase Farmacocinética y farmacodinamia). Medicamentos como tamoxifeno y otros tratamientos que contengan estrógenos no deben ser coadministrados con ARIMIDEX® debido a que disminuyen su acción farmacológica.
Alteraciones en los resultados de pruebas de laboratorio: Con poca frecuencia (≥0.1% y < 1%) se ha reportado elevación de gama glutamiltransferasa (gama-GT) y de bilirrubina.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Toxicidad aguda: En estudios con roedores la dosis letal mediana de anastrazol fue mayor de 100 mg/kg por vía oral y mayor de 50 mg/kg por vía intraperitoneal. En un estudio de toxicidad aguda en perros por vía oral, la mediana de la dosis letal fue mayor de 45 mg/kg. Toxicidad crónica: Se realizaron múltiples estudios de toxicidad en ratas y perros y no se establecieron niveles sin efecto en estos estudios de toxicidad con anastrozol, pero aquellos efectos observados a dosis baja (1mg/kg/día) y dosis media (perro 3 mg/kg/día; rata 5 mg/kg/día) estuvieron relacionados con las propiedades ya sea farmacológicas o bien inductoras de enzimas de anastrozol y no estuvieron acompañados de cambios tóxicos o degenerativos significativos. Mutagenicidad: Los estudios de toxicología genética con anastrozol muestran que no es mutagénico ni clastogénico. Toxicología reproductiva: La administración oral de anastrozol en ratas y conejas preñadas no produjo efectos teratogénicos con dosis de hasta 1.0 y 0.2 mg/kg/día respectivamente. Efectos observados (aumento del tamaño de la placenta en ratas y embarazos mal logrados en conejas) estuvieron asociados con la farmacología del producto. La administración oral de anastrozol en ratas hembra produjo una alta incidencia de infertilidad con 1 mg/kg/día así como un incremento en la pérdida de pre-implantación con 0.02 mg/kg/día. Estos efectos estuvieron relacionados con la farmacología del compuesto y fueron revertidos completamente después de un periodo de suspensión del producto de cinco semanas. La supervivencia de las camadas de ratas a las que se les administró anastrozol 0.02 mg/kg/día y más (desde el día 17 de gestación hasta el día 22 post-parto) estuvo comprometida. Estos efectos se relacionaron con los efectos farmacológicos del compuesto durante el parto. No hubo efectos adversos en el comportamiento o en la función reproductiva de la primera generación de críos atribuible al tratamiento materno con anastrozol. Carcinogenicidad: Resultados de dos años de estudios sobre oncogenicidad en ratas mostraron un aumento en la incidencia de tumores hepáticos, pólipos uterinos en hembras y adenomas tiroideos en machos solamente a dosis elevadas (25 mg/kg/día). Estos cambios ocurrieron con dosis que representan 100 veces las dosis terapéuticas en humanos y no son considerados clínicamente significativos en pacientes tratados con anastrozol. Resultados de dos años de estudios sobre oncogenicidad en ratones tuvieron como resultado la inducción de tumores benignos ováricos y variaciones en la incidencia de tumores linforreticulares (menos sarcomas histiocíticos en hembras y más muertes como resultado de linfomas). Estos estudios están considerados como efectos específicos en ratones por la inhibición de aromatasa y no son clínicamente significativos en pacientes tratados con anastrozol.
Dosis y vía de administración: Adultos (incluyendo pacientes de edad avanzada): Una tableta de 1 mg al día, por vía oral. Niños: No se recomienda usar en niños, ya que su eficacia no ha sido establecida (Véase Farmacocinética y farmacodinamia) Insuficiencia renal: No se recomienda modificar la dosis. Insuficiencia hepática: No se recomienda modificar la dosis.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: Existe experiencia clínica limitada en relación a sobredosis de ARIMIDEX®. No hay reportes de que algún paciente haya tomado una dosis que exceda los 60 mg. No se ha observado toxicidad ni tampoco se ha visto ningún efecto clínico relevante. Se observó toxicidad aguda en animales con una dosis mayor de 45 mg/kg (equivalente a 2.7 g). Se han realizado estudios clínicos con dosis únicas de hasta 60 mg en voluntarios sanos hombres y hasta 10 mg diarios en mujeres postmenopáusicas con cáncer de mama avanzado; ambas dosis resultaron bien toleradas. No se ha establecido una dosis única de ARIMIDEX® que produzca síntomas que pongan en peligro la vida. No existe un antídoto específico para la sobredosis y su tratamiento debe ser sintomático. Durante el manejo de una sobredosis debe considerarse la posibilidad de que múltiples agentes hayan sido ingeridos. El vómito puede inducirse si el paciente se encuentra consciente. La diálisis puede ser útil debido a que ARIMIDEX® no se une fuertemente a proteínas. Cuidados generales, incluyendo la vigilancia frecuente de los signos vitales y la observación cercana del paciente, son medidas indicadas.
Presentación(es): Caja con 14 tabletas con 1 mg en envase de burbuja. Caja con 28 tabletas con 1 mg en envase de burbuja.
Recomendaciones sobre almacenamiento: Consérvese a temperatura ambiente a no más de 30°C y en lugar seco.
Leyendas de protección: Su venta requiere receta médica. No se deje al alcance de los niños. No se use en el embarazo ni en la lactancia. Este medicamento deberá ser administrado únicamente por médicos especialistas en Oncología y con experiencia en quimioterapia antineoplásica. Literatura exclusiva para médicos. Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx
Nombre y domicilio del laboratorio: AstraZeneca, S.A. de C.V. Super Av. Lomas Verdes No.67, Fracc. Lomas Verdes, C.P. 53120.Naucalpan de Juárez, México.
Número de registro del medicamento: 263M98 SSA IV.