ATRIPLA®
STENDHAL
Denominación genérica: Efavirenz, emtricitabina y tenofovir disoproxil fumarato.
Forma farmacéutica y formulación: Tabletas recubiertas composición: Cada tableta contiene: Efavirenz 600 mg, Emtricitabina 200 mg, Tenofovir disoproxil fumarato 300 mg (equivalente a 245 mg de tenofovir disoproxil). Excipientes c.b.p. 1 tableta recubierta.
Indicaciones terapéuticas: ATRIPLA® está indicado para su uso solo como esquema terapéutico completo o en asociación con otros antirretrovirales, para el tratamiento de la infección por el virus de la inmunodeficiencia humana de tipo 1 (VIH-1) en los adultos.
Farmacocinética y farmacodinamia: Estudios clínicos: El estudio clínico 934 respalda el uso de las tabletas de ATRIPLA® en los pacientes infectados por el VIH-1 sin tratamiento previo con antirretrovirales. Puede encontrarse información adicional en respaldo del uso de ATRIPLA® en los pacientes sin tratamiento previo con antirretrovirales en la ficha técnica de viread. El estudio clínico 073 proporciona experiencia clínica sobre sujetos con supresión virológica y sin antecedentes de fracaso virológico, quienes cambiaron su tratamiento actual por ATRIPLA®. En los pacientes con tratamiento previo con antirretrovirales, el uso de las tabletas de ATRIPLA® puede plantearse en los sujetos con cepas del VIH-1 que se espera que sean sensibles a los componentes de ATRIPLA®, según la evaluación de los antecedentes de tratamiento o mediante pruebas genotípicas o fenotípicas [Véase Farmacología clínica]. Estudio 934: Se informan los datos obtenidos a 144 semanas en el estudio 934, un ensayo multicéntrico, aleatorizado, abierto y con testigo activo, en el que se comparó emtricitabina + tenofovir DF, administrados en asociación con efavirenz, frente a la asociación de dosis fijas de zidovudina y lamivudina, en asociación con efavirenz, en 511 sujetos sin tratamiento previo con antirretrovirales. Desde la semana 96 a la semana 144 del ensayo, los sujetos recibieron una asociación de dosis fijas de emtricitabina y tenofovir DF con efavirenz, en lugar de emtricitabina + tenofovir DF con efavirenz. Los sujetos tenían una media de edad de 38 años (intervalo de 18 a 60), el 86% eran varones; el 59%, de raza blanca, y el 23% eran negros. El recuento inicial medio de linfocitos CD4+ fue de 245 linfocitos/mm3 (intervalo de 2 a 1191), y la mediana del ARN del VIH-1 plasmático inicial fue de 5.01 log10 copias/ml (intervalo de 3.56 a 6.54). Los sujetos se estratificaron según el recuento inicial de linfocitos CD4+ ( < o ≥ 200 linfocitos/mm3); el 41% tenía recuentos de linfocitos CD4+ de < 200 linfocitos/mm3. El 51% de los sujetos tenía cargas virales basales de > 100,000 copias/ml. En la tabla 1 se presentan los resultados del tratamiento después de 48 y de 144 semanas en los sujetos que no presentaban resistencia al efavirenz al inicio (n = 487). Tabla 1. Resultados del tratamiento asignado aleatoriamente en las semanas 48 y 144 (estudio 934).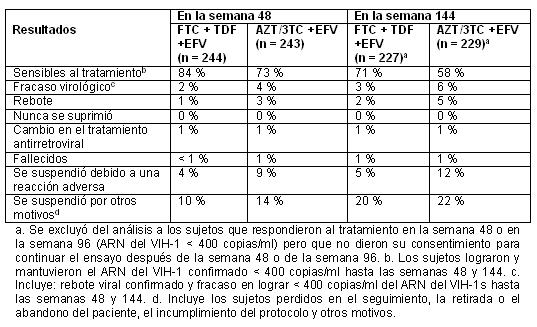
Hasta la semana 48, el 84% de los sujetos del grupo tratado con emtricitabina + tenofovir DF y el 73% de los sujetos tratados con la asociación de zidovudina y lamivudina lograron y mantuvieron el ARN del VIH-1 < 400 copias/ml (hasta la semana 144: 71% y 58%, respectivamente). En este ensayo abierto, la diferencia en la proporción de sujetos que lograron y mantuvieron el ARN del VIH-1 < 400 copias/ml después de 48 semanas de tratamiento, es principalmente el resultado del mayor número de interrupciones del tratamiento debidas a reacciones adversas y a otros motivos en el grupo tratado con la asociación de zidovudina y lamivudina. Además, el 80% de los sujetos del grupo tratado con emtricitabina + tenofovir DF y el 70% de los sujetos tratados con la asociación de zidovudina y lamivudina lograron y mantuvieron el ARN del VIH-1 < 50 copias /ml hasta la semana 48 (hasta la semana 144: 64% y 56%, respectivamente). En la semana 48, el aumento medio, respecto de los valores iniciales del recuento de linfocitos CD4+ fue de 190 linfocitos/mm3 en el grupo tratado con emtricitabina + tenofovir DF, y de 158 linfocitos/mm3 en el grupo que recibió la asociación de zidovudina y lamivudina (en la semana 144: 312 y 271 linfocitos/mm3, respectivamente). A las 48 semanas, siete sujetos del grupo tratado con emtricitabina + tenofovir DF y cinco sujetos del grupo tratado con la asociación de zidovudina y lamivudina presentaron una nueva reacción de clase C, según el código de los CDC (diez y seis sujetos, respectivamente, hasta las 144 semanas). Estudio 073: El estudio 073 fue un ensayo clínico abierto y con asignación aleatoria, de 48 semanas de duración, en sujetos con supresión virológica estable, que recibían tratamiento antirretroviral asociado que consistía en al menos dos inhibidores de la transcriptasa reversa análogos a nucleósidos (ITRAN) administrados en asociación con un inhibidor de la proteasa (con o sin ritonavir) (INNRT). Para incluirse en el estudio, los sujetos debían tener un valor de ARN del VIH-1 < 200 copias/ml durante al menos 12 semanas con su tratamiento actual, sin mutaciones conocidas del VIH-1 que confiriesen resistencia a los componentes de ATRIPLA® ni antecedentes de falla virológica. El ensayo comparó la eficacia de cambiar el tratamiento por ATRIPLA® o de continuar con el tratamiento antirretroviral basal (EB) Se asignó aleatoriamente a los sujetos, en una proporción de 2:1, para cambiar a ATRIPLA® (n = 203) o continuar en su esquema basal (EB) (n = 97). Los sujetos tenían una edad media de 43 años (límites, 22 y 73 años); el 88% eran varones; el 68%, de raza blanca; el 29%, negros o afroamericanos; y el 3%, de otras razas. Al inicio, la mediana del recuento de linfocitos CD4+ fue de 516 linfocitos/mm3 y el 96% tenía un valor de ARN del VIH-1 < 50 copias/ml. La mediana del tiempo desde el inicio del tratamiento antirretroviral fue de tres años y el 88% de los sujetos estaba recibiendo su primer tratamiento antirretroviral en el momento de la inscripción en el ensayo. En la semana 48, el 89% y el 87% de los sujetos que cambiaron a Atripla® mantuvieron un ARN del VIH de < 200 copias/ml y de < 50 copias/ml, respectivamente, en comparación con el 88% y el 85% de los sujetos que continuaron con su EB; esta diferencia no fue estadísticamente significativa. No se observaron cambios en la cuenta de linfocitos CD4+ entre el inicio y la semana 48 en ninguno de los grupos de tratamiento. Descripción: ATRIPLA® es una asociación de dosis fija, en tabletas, que contiene efavirenz, emtricitabina y tenofovir disoproxil fumarato (tenofovir DF). STOCRIN es el nombre comercial de efavirenz, un inhibidor de la transcriptasa reversa no análogo nucleósido. Emtriva es el nombre comercial de emtricitabina, un análogo nucleosídico sintético de la citidina. Viread es el nombre comercial del FD tenofovir, que se convierte in vivo en tenofovir, un análogo fosfonato nucleosídico acíclico (nucleotídico) del 5'-monofosfato de adenosina. Viread y Emtriva son los componentes de Truvada. Las tabletas de ATRIPLA® son para administración oral. Cada comprimido contiene 600 mg de efavirenz, 200 mg de emtricitabina y 300 mg de tenofovir disoproxil fumarato (que es equivalente a 245 mg tenofovir disoproxilo), como principios activos. Las tabletas contienen los siguientes componentes inactivos: croscarmelosa sódica, hidroxipropilcelulosa, estearato de magnesio, celulosa microcristalina y laurilsulfato de sodio. Las tabletas están recubiertas con película, con un material que contiene óxido de hierro negro, polietilenglicol, alcohol polivinílico, óxido de hierro rojo, talco y dióxido de titanio. Efavirenz: El nombre químico del efavirenz es (S)-6-cloro-4-(ciclopropiletinil)-1,4-dihidro-4(trifluorometil)-2H-3,1-benzoxazin-2-ona. Su fórmula molecular es C14H9ClF3NO2 y su fórmula estructural es: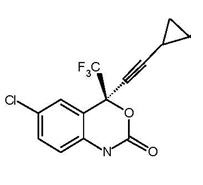
El efavirenz es un polvo cristalino, de color entre blanco y ligeramente rosado, con una masa molecular de 315.68. Es prácticamente no hidrosoluble (menos de 10 mg/ml). Emtricitabina: El nombre químico de la emtricitabina es 5-fluoro-1-(2R, 5S)-[2(hidroximetil)-1,3-oxatiolan-5-il] citosina. La emtricitabina es el enantiómero (-) de un análogo tio de la citidina, que difiere de otros análogos de la citidina porque tiene un flúor en la posición 5. Su fórmula molecular es C8H10FN3O3S y su peso molecular es 247.24. Tiene la siguiente fórmula estructural: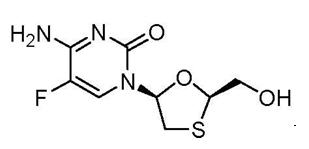
La emtricitabina es un polvo cristalino, de color entre blanco y blanquecino, con una hidrosolubilidad de aproximadamente 112 mg/ml, a 25°C. Tenofovir disoproxil fumarato: El tenofovir disoproxil fumarato es una sal del ácido fumárico del derivado éster bis-isopropoxicarboniloximetil del tenofovir. El nombre químico del tenofovir disoproxil fumarato es fumarato de 9-[(R)-2[[bis[[(isopropoxicarbonil)oxi]-metoxi]fosfinil]metoxi]propil]adenina (1:1). Su fórmula molecular es C19H30N5O10P • C4H4O4 y su peso molecular es 635.52. Tiene la siguiente fórmula estructural: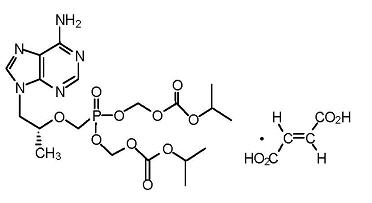
El tenofovir disoproxil fumarato es un polvo cristalino, de color entre blanco y blanquecino, con una hidrosolubilidad de aproximadamente 13.4 mg/ml a 25°C. Microbiología: Mecanismo de acción: Efavirenz: El efavirenz es un inhibidor de la transcriptasa reversa no análogo nucleósido (ITRNN) del VIH-1. La actividad del efavirenz está mediada predominantemente por la inhibición no competitiva de la transcriptasa reversa del VIH-1. El efavirenz no inhibe la transcriptasa reversa del VIH-2 ni las polimerasas a, b, c y d del ADN de las células humanas. Emtricitabina: La emtricitabina, un análogo nucleosídico sintético de la citidina, es fosforilada por las enzimas celulares para formar 5'-trifosfato de emtricitabina. El 5'-trifosfato de emtricitabina inhibe la actividad de la transcriptasa reversa del VIH-1 al competir con el sustrato natural 5'-trifosfato de desoxicitidina e incorporarse en el ADN viral incipiente, lo que produce la terminación de la cadena. El 5'-trifosfato de emtricitabina es un inhibidor débil de las polimerasas a, b y e del ADN de los mamíferos, y de la polimerasa c del ADN mitocondrial. Tenofovir disoproxil fumarato: El tenofovir disoproxil fumarato es un análogo diéster de fosfonato nucleosídico acíclico del monofosfato de adenosina. El tenofovir DF requiere la hidrólisis inicial del diéster para su conversión en tenofovir y fosforilaciones posteriores por las enzimas celulares para formar el difosfato de tenofovir. El difosfato de tenofovir inhibe la actividad de la transcriptasa reversa del VIH-1 al competir con el sustrato natural 5'-trifosfato de desoxiadenosina y, después de su incorporación en el ADN, al terminar la cadena de ADN. El difosfato de tenofovir es un inhibidor débil de las polimerasas a y b del ADN de los mamíferos, y de la polimerasa c del ADN mitocondrial. Actividad antiviral: Efavirenz, emtricitabina y tenofovir disoproxil fumarato: En estudios de asociaciones en los que se evaluó la actividad antiviral en cultivos de células de la emtricitabina y el efavirenz juntos, el efavirenz y el tenofovir juntos, y la emtricitabina y el tenofovir juntos, se observaron efectos antivirales entre aditivos y sinérgicos. Efavirenz: La concentración de efavirenz que inhibe la replicación de las cepas no mutantes adaptadas en el laboratorio y cepas aisladas clínicas en cultivos de células entre el 90 y el 95% (CE90-95) varió entre 1.7 y 25 nM en cultivos de líneas celulares linfoblastoides, leucocitos mononucleares en la sangre periférica y de macrófagos o monocitos. El efavirenz demostró una actividad antiviral aditiva contra el VIH-1, en cultivos de células, al asociarse a inhibidores de la transcriptasa reversa no análogos nucleósidos(ITRNN) (delavirdina y nevirapina), inhibidores de la transcriptasa reversa análogos a nucleósidos (ITRAN) (abacavir, didanosina, lamivudina, estavudina, zalcitabina y zidovudina), inhibidores de la proteasa (IP) (amprenavir, indinavir, lopinavir, nelfinavir, ritonavir y saquinavir), y el inhibidor de la fusión, enfuvirtida. El efavirenz demostró una actividad antiviral entre aditiva y antagonista en cultivo de células con el atazanavir. El efavirenz demostró una actividad antiviral contra el clade B y la mayoría de los aislados que no son del clade B (subtipos A, AE, AG, C, D, F, G, J y N), pero tuvo una actividad antiviral reducida contra los virus del grupo O. El efavirenz no es activo contra el VIH-2. Emtricitabina: Se evaluó la actividad antiviral de la emtricitabina en cultivo de células contra cepas aisladas clínicas y de laboratorio del VIH-1 en líneas celulares linfoblastoides, la línea de células MAGI-CCR5 y leucocitos mononucleares en la sangre periférica. Los valores de la concentración eficaz al 50% (CE50) de la emtricitabina estuvieron entre los límites de 0.0013 y 0.64 mM (de 0.0003 a 0.158 mg/ml). En estudios de asociaciones farmacológicas de la emtricitabina con inhibidores de la transcriptasa reversa análogos a nucléosidos (abacavir, lamivudina, estavudina, zalcitabina y zidovudina), inhibidores de la transcriptasa reversa no análogos nucleósidos (delavirdina, efavirenz y nevirapina) e inhibidores de la proteasa (amprenavir, nelfinavir, ritonavir y saquinavir) se observaron efectos entre aditivos y sinérgicos. La emtricitabina presentó actividad antiviral en cultivo de células contra los clades A, B, C, D, E, F y G del VIH-1 (los valores de CE50 estuvieron entre los límites de 0.007 y 0.075 mM), y mostró actividad específica de cepas contra el VIH-2 (los valores de CE50 estuvieron entre los límites de 0.007 y 1.5 mM). Tenofovir disoproxil fumarato: Se evaluó la actividad antiviral en cultivo de células del tenofovir contra cepas aisladas clínicas y de laboratorio del VIH-1 en líneas celulares linfoblastoides, células primarias de monocitos/macrófagos y linfocitos en la sangre periférica. Los valores de CE50 correspondientes al tenofovir estuvieron dentro de los límites de 0.04 y 8.5 mM. En estudios de asociaciones farmacológicas del tenofovir con inhibidores de la transcriptasa reversa análogos nucleósidos (abacavir, didanosina, lamivudina, estavudina, zalcitabina y zidovudina), inhibidores de la transcriptasa reversa no análogos nucleósidos (delavirdina, efavirenz y nevirapina) e inhibidores de la proteasa (amprenavir, indinavir, nelfinavir, ritonavir y saquinavir) se observaron efectos entre aditivos y sinérgicos. El tenofovir presentó actividad antiviral en cultivo de células contra los clades A, B, C, D, E, F, G y O del VIH-1 (los valores de CE50 variaron entre 0.5 y 2.2 mM), y mostró actividad específica de cepas contra el VIH-2 (los valores de CE50 variaron entre 1.6 mM y 5.5 mM). Efavirenz, emtricitabina y tenofovir disoproxil fumarato: Se han seleccionado en cultivos de células y en ensayos clínicos cepas aisladas del VIH-1 con disminución de la sensibilidad a la asociación de emtricitabina y tenofovir. El análisis genotípico de estas cepas aisladas identificó las sustituciones de aminoácidos M184V/I o K65R en la TR viral. En un ensayo clínico con sujetos sin tratamiento antirretroviral previo [estudio 934; véase Farmacocinética y Farmacodinamia] se realizó un análisis de resistencia en cepas aisladas del VIH-1 de todos los sujetos con falla virológica confirmada que tenían más de 400 copias/ml de ARN del VIH-1 en la semana 144 o que habían abandonado prematuramente el estudio. La forma más frecuente de resistencia que se produjo fue la resistencia genotípica al efavirenz, predominantemente la sustitución K103N. La resistencia al efavirenz se produjo en 13/19 sujetos analizados del grupo tratado con emtricitabina + tenofovir DF, y en 21/29 sujetos analizados del grupo que recibió la dosis fijas de zidovudina y lamivudina. Se observó la sustitución de aminoácidos M184V, asociada con resistencia a la emtricitabina y la lamivudina, en 2/19 de las cepas aisladas de los sujetos analizados del grupo tratado con emtricitabina + tenofovir FD y en 10/29 de las cepas aisladas de los sujetos analizados del grupo tratado con la asociación de zidovudina y lamivudina. En las 144 semanas del estudio 934, ningún sujeto presentó una sustitución K65R detectable en su VIH-1, según los análisis genotípicos habituales. En un ensayo clínico en sujetos sin tratamiento antirretroviral previo, las cepas aisladas de 8/47 (17%) sujetos analizados, que recibían tenofovir DF, presentaron la sustitución K65R hasta las 144 semanas de tratamiento; siete de estos casos se produjeron en las 48 primeras semanas de tratamiento y uno en la semana 96. En los sujetos con tratamiento antirretroviral previo, 14/304 (5%) de los sujetos tratados con tenofovir DF y con falla virológica hasta la semana 96 presentaron una disminución de la sensibilidad de más de 1.4 veces mayor (mediana, 2.7) al tenofovir. El análisis genotípico de los las cepas aisladas resistentes mostró una sustitución en el gen de la TR del VIH-1, que produjo la sustitución de aminoácido K65R. Efavirenz: Se han obtenido cepas aisladas clínicas con una disminución de la sensibilidad al efavirenz en cultivo de células. La sustitución de aminoácido observada con mayor frecuencia en los ensayos clínicos con efavirenz es K103N (54%). En sujetos que no respondieron al tratamiento con efavirenz asociado a otros antirretrovirales, se observaron una o más sustituciones de la transcriptasa reversa en las posiciones de los aminoácidos 98, 100, 101, 103, 106, 108, 188, 190, 225, 227 y 230. Otras sustituciones de resistencia que se observó que aparecieron con frecuencia son L100 I (7%), K101E/Q/R (14%), V108I (11%), G190S/T/A (7%), P225H (18%) y M230I/L (11%). En una selección en cultivo de células, aparecieron rápidamente cepas aisladas del VIH-1 con una disminución de la sensibilidad al efavirenz (aumento más de 380 veces mayor del valor de CE90). La caracterización genotípica de estos virus identificó sustituciones de un solo aminoácido L100I o V179D, sustituciones dobles L100I/V108I y sustituciones triples L100I/V179D/Y181C de la transcriptasa reversa. Emtricitabina: Se han seleccionado cepas aisladas del VIH-1 resistentes a la emtricitabina en cultivos de células y en ensayos clínicos. El análisis genotípico de estas cepas aisladas demostró que la disminución de la sensibilidad a la emtricitabina se asoció a una sustitución del gen de la retrotranscriptasa del VIH-1 en el codón 184, que produjo una sustitución del aminoácido metionina por valina o isoleucina (M184V/I). Tenofovir disoproxil fumarato: En un cultivo de células se seleccionaron cepas aisladas del VIH-1 con una disminución de la sensibilidad al tenofovir. Estos virus expresaron una sustitución K65R en la transcriptasa reversa y mostraron una reducción del doble al cuádruple de la sensibilidad al tenofovir. Resistencia cruzada: Efavirenz, emtricitabina y tenofovir disoproxil fumarato: Se ha reconocido la resistencia cruzada entre inhibidores de la transcriptasa reversa no análogos a nucleósidos. Se ha reconocido también la resistencia cruzada entre ciertos inhibidores de la transcriptasa reversa análogos a nucleósidos. Las sustituciones M184V/I o K65R seleccionadas en el cultivo de células por la asociación de emtricitabina y tenofovir también se observan en algunas cepas aisladas del VIH-1 de sujetos que no responden al tratamiento con tenofovir en asociación con lamivudina o emtricitabina, y abacavir o didanosina. Por lo tanto, la resistencia cruzada entre estos fármacos puede presentarse en los pacientes cuyos virus presentan alguna de estas sustituciones de aminoácidos o ambas. Efavirenz: Las cepas aisladas clínicas que se caracterizaron anteriormente como resistentes al efavirenz también fueron fenotípicamente resistentes, en cultivo de células, a la delavirdina y la nevirapina, en comparación con los valores iniciales. Los aislados virales clínicos, resistentes a la delavirdina o a la nevirapina, con sustituciones asociadas a la resistencia a los ITRNN (A98G, L100I, K101E/P, K103N/S, V106A, Y181X, Y188X, G190X, P225H, F227L o M230L) mostraron una disminución de la sensibilidad al efavirenz en el cultivo de células. Más del 90% de las cepas aisladas resistentes a los IITRAN examinadas en el cultivo de células conservaron la sensibilidad al efavirenz. Emtricitabina: Las cepas aisladas resistentes a la emtricitabina (M184V/I) presentaron resistencia cruzada a la lamivudina y la zalcitabina; sin embargo, en el cultivo de células, conservaron la sensibilidad a la didanosina, la estavudina, el tenofovir, la zidovudina y los ITRNN (delavirdina, efavirenz y nevirapina). Las cepas aisladas de VIH-1 que contienen la sustitución K65R, seleccionadas in vivo por el abacavir, la didanosina, el tenofovir y la zalcitabina, presentaron una disminución de la sensibilidad a la inhibición por la emtricitabina. Los virus que presentan sustituciones que confieren una disminución de la sensibilidad a la estavudina y la zidovudina (M41L, D67N, K70R, L210W, T215Y/F y K219Q/E), o a la didanosina (L74V) se mantuvieron sensibles a la emtricitabina. Tenofovir disoproxil fumarato: La sustitución K65R seleccionada por el tenofovir también se selecciona en algunos pacientes infectados por el VIH-1, tratados con abacavir, didanosina o zalcitabina. Las cepas aisladas del VIH-1 con esta sustitución también mostraron una disminución de la sensibilidad a la emtricitabina y la lamivudina. Por lo tanto, puede presentarse resistencia cruzada entre estos fármacos en los pacientes cuyos virus hospedan la sustitución K65R. Las cepas aisladas del VIH-1 de sujetos (n = 20) cuyo VIH-1 expresó una media de tres sustituciones de aminoácidos de la TR asociadas a la zidovudina (M41L, D67N, K70R, L210W, T215Y/F o K219Q/E/N) presentaron una disminución de 3.1 veces de la sensibilidad al tenofovir. Los sujetos cuyo virus expresó una sustitución L74V sin sustituciones asociadas con la resistencia a la zidovudina (n = 8) tuvieron una respuesta reducida a VIREAD. Se dispone de datos limitados sobre los pacientes cuyo virus expresó una sustitución Y115F (n = 3), una sustitución Q151M (n = 2) o una inserción T69 (n = 4); todos ellos tuvieron una respuesta reducida. Farmacología clínica. Farmacocinética: ATRIPLA®: Una tableta recubierta de ATRIPLA® es bioequivalente a un comprimido de STOCRIN (600 mg), más una cápsula de EMTRIVA (200 mg), más un comprimido de VIREAD (300 mg) después de su administración como dosis única a sujetos sanos en ayunas (n = 45). Efavirenz: En los sujetos infectados por el VIH-1, el tiempo hasta alcanzar las concentraciones máximas en el plasma fue de aproximadamente tres a cinco horas, y las concentraciones plasmáticas en el estado de equilibrio se alcanzaron después de seis a diez días. En 35 sujetos infectados por el VIH-1 que recibieron 600 mg de efavirenz una vez al día, la Cmáx en el estado de equilibrio fue de 12.9 ± 3.7 mM (media ± DE); la Cmín fue de 5.6 ± 3.2 mM, y el AUC, 184 ± 73 mM.h. El efavirenz presenta una elevada fijación a las proteínas del plasma humano (aproximadamente entre el 99.5 y el 99.75%), predominantemente a la albúmina. Después de la administración de efavirenz marcado con 14C, se recuperó del 14 al 34% de la dosis en la orina (principalmente como metabolitos), y se recuperó entre el 16 y el 61% en las heces (principalmente como el fármaco original). En los estudios in vitro se sugiere que las principales isoenzimas responsables del metabolismo del efavirenz son CYP3A y CYP2B6. Se ha demostrado que el efavirenz induce las enzimas del CYP, lo que produce la inducción de su propio metabolismo. El efavirenz tiene una semivida terminal de 52 a 76 horas después de la administración de dosis únicas, y de 40 a 55 horas después de la administración de dosis múltiples. Emtricitabina: La emtricitabina se absorbe de manera rápida después de la administración por vía oral, y se alcanzan concentraciones plasmáticas máximas entre una y dos horas después de la dosis. Después de la administración por vía oral de dosis múltiples de emtricitabina a 20 sujetos infectados por el VIH-1, la concentración máxima (Cmáx) de emtricitabina en el plasma, en el estado de equilibrio, fue de 1.8 ± 0.7 mg/ml (media ± DE), y el AUC durante un intervalo de dosificación de 24 horas fue de 10.0 ± 3.1 mg.h/ml. La concentración mínima media en el plasma, en el estado de equilibrio, 24 horas después de administrada la dosis, fue de 0.09 mg/ml. La biodisponibilidad absoluta media de la emtricitabina fue del 93%. La unión in vitro de la emtricitabina a las proteínas plasmáticas en los seres humanos es inferior al 4% y es independiente de la concentración, entre los límites de 0.02 y 200 mg/ml. Después de la administración de emtricitabina radiomarcada, se recupera aproximadamente el 86% en la orina y el 13% como metabolitos. Los metabolitos de la emtricitabina son 3'-sulfóxido diastereómeros y su conjugado con ácido glucurónico. La emtricitabina se elimina mediante una combinación de filtración glomerular y secreción tubular activa, con una depuración renal en los adultos con función renal normal de 213 ± 89 ml/min (media ± DE). Después de una dosis única por vía oral, la semivida plasmática de la emtricitabina es de aproximadamente 10 horas. Tenofovir disoproxil fumarato: Después de la administración por vía oral de una dosis única de 300 mg de tenofovir DF a sujetos infectados por el VIH-1 en ayunas, las concentraciones séricas máximas (Cmáx) se alcanzaron en 1.0 ± 0.4 horas (media ± DE). Los valores de la Cmáx y el AUC fueron de 296 ± 90 ng/ml y 2287 ± 685 ng•h/ml, respectivamente. La biodisponibilidad oral del tenofovir a partir del tenofovir DF en los sujetos en ayunas es de aproximadamente el 25%. La unión in vitro del tenofovir a las proteínas plasmáticas en los seres humanos es inferior al 0.7% y es independiente de la concentración, dentro de los límites de 0.01 y 25 mg/ml. Aproximadamente entre el 70 y el 80% de la dosis de tenofovir administrada por vía intravenosa se recupera como fármaco inalterado en la orina. El tenofovir se elimina mediante una combinación de filtración glomerular y secreción tubular activa, con una depuración renal en los adultos con función renal normal de 243 ± 33 ml/min (media ± DE). Después de una dosis oral única de tenofovir, la semivida de eliminación terminal es de aproximadamente 17 horas. Efectos de los alimentos en la absorción oral: No se ha evaluado ATRIPLA® en presencia de alimentos. La administración de tabletas de efavirenz con una comida rica en grasas aumentó el AUC media del efavirenz en un 28%, y la Cmáx media, en un 79%, en comparación con su administración en ayunas. Comparado con la administración en ayunas, la administración de la combinación de tenofovir DF y emtricitabina, con una comida rica en grasas o una comida ligera, aumentó el AUC media en un 35%, y la Cmáx, en un 15%, sin ningún efecto sobre la exposición a la emtricitabina [Dosis y via de administración y Precauciones generales, Información para los pacientes]. Poblaciones especiales: Raza: Efavirenz: Las características farmacocinéticas del efavirenz en los sujetos infectados por el VIH-1 parecen ser similares entre los grupos raciales estudiados. Emtricitabina: No se han identificado diferencias farmacocinéticas debidas a la raza después de la administración de emtricitabina. Tenofovir disoproxil fumarato: No hubo un número suficiente de sujetos de grupos raciales y étnicos, aparte de la raza blanca, para poder determinar adecuadamente las posibles diferencias farmacocinéticas entre estas poblaciones después de la administración de tenofovir DF. Sexo: Efavirenz, emtricitabina y tenofovir disoproxil fumarato: Las propiedades farmacocinéticas del efavirenz, la emtricitabina y el tenofovir son parecidas en los sujetos de ambos sexos. Pacientes pediátricos y geriátricos: No se han realizado ensayos farmacocinéticos del tenofovir DF en los sujetos pediátricos (menores de 18 años). No se ha estudiado el efavirenz en los sujetos pediátricos menores de tres años o con un peso inferior a 13 kg. Se ha estudiado la emtricitabina en los sujetos pediátricos de tres meses a 17 años. No se recomienda la administración de ATRIPLA® a los sujetos pediátricos. No se han evaluado completamente las características farmacocinéticas del efavirenz, la emtricitabina y el tenofovir en los ancianos (mayores de 65 años). [Dosis y vía de administración]. Pacientes con falla renal: Efavirenz: No se han estudiado las características farmacocinéticas del efavirenz en los sujetos con insuficiencia renal; sin embargo, menos del 1% del efavirenz se excreta inalterado en la orina, por lo que la repercusión en la disfunción renal sobre la eliminación del efavirenz debería ser mínima. Emtricitabina y tenofovir disoproxil fumarato: Las propiedades farmacocinéticas de la emtricitabina y del tenofovir DF están alteradas en los sujetos con disfunción renal. En los sujetos con una depuración de creatinina inferior a 50 ml/min, la Cmáx y el AUC0-∞ de la emtricitabina y el tenofovir aumentaron [Precauciones generales]. Pacientes con falla hepática. Efavirenz: Un ensayo con dosis múltiples mostró que no se produce ningún efecto significativo en las características farmacocinéticas del efavirenz en sujetos con disfunción hepática leve (clase A de Child-Pugh), en comparación con los controles. No hubo datos suficientes para determinar si la falla hepática moderada o grave (clase B o C de Child-Pugh) afecta las características farmacocinéticas del efavirenz [Precauciones generales]. Emtricitabina: No se han estudiado las características farmacocinéticas de la emtricitabina en los sujetos con falla hepática; sin embargo, las enzimas hepáticas no metabolizan significativamente la emtricitabina, por lo que la repercusión de la disfunción hepática debería ser limitada. Tenofovir disoproxil fumarato: Se han estudiado las características farmacocinéticas del tenofovir después de una dosis de 300 mg de tenofovir DF en sujetos no infectados por el VIH y con falla hepática de moderada a grave. No hubo alteraciones importantes en las propiedades farmacocinéticas del tenofovir en sujetos con disfunción hepática, en comparación con los sujetos con función hepática normal.
Interacciones medicamentosas y de otro género: Los ensayos de interacción medicamentosa descritos se efectuaron con efavirenz, emtricitabina o tenofovir DF como fármacos individuales; no se han realizado ensayoss de interacción medicamentosa con ATRIPLA®. Efavirenz: Las características farmacocinéticas del efavirenz y el tenofovir en el estado de equilibrio no se vieron afectadas cuando se administraron efavirenz y tenofovir disoproxil fumarato juntos, en comparación con la administración de cada fármaco por separado. No se han realizado ensayos específicos de interacción medicamentosa con efavirenz e ITRAN aparte del tenofovir, la lamivudina y la zidovudina. No se espera que se produzcan interacciones clínicamente significativas basadas en las vías de eliminación de los ITRAN.Se ha demostrado que el efavirenz causa, in vivo, inducción de las enzimas hepáticas, aumentando así la biotransformación de algunos fármacos metabolizados por el CYP3A. En estudios in vitro, se ha demostrado que el efavirenz inhibió las isoenzimas 2C9, 2C19 y 3A4 del CYP, con valores de Ki (entre 8.5 y 17 mM) dentro de los límites de las concentraciones plasmáticas observadas de efavirenz. En estudios in vitro, el efavirenz no inhibió el CYP2E1, e inhibió el CYP2D6 y el CYP1A2 (valores de Ki entre 82 y 160 mM), sólo en concentraciones muy superiores a las alcanzadas clínicamente. La administración concomitante de efavirenz con fármacos que son metabolizados principalmente por las isoenzimas 2C9, 2C19 y 3A4 puede producir una alteración de las concentraciones plasmáticas del fármaco administrado concomitantemente. Se esperaría que los fármacos que inducen la actividad del CYP3A4 aumenten la depuración del efavirenz, produciéndose una disminución de las concentraciones plasmáticas. Se realizaron ensayos de interacción medicamentosa con efavirenz y otros fármacos con probabilidad de ser administrados concomitantemente, o con fármacos usados con frecuencia como sondas para la interacción farmacocinética. No se observó ninguna interacción clínicamente significativa entre el efavirenz y la zidovudina, la lamivudina, la azitromicina, el fluconazol, el lorazepam, la cetirizina ni la paroxetina. Las dosis únicas de famotidina o de un antiácido de aluminio y magnesio con simeticona no tuvieron ningún efecto sobre las exposiciones del efavirenz. Los efectos de la administración concomitante de efavirenz sobre la Cmáx, el AUC y la Cmín se resumen en la tabla 2 (efecto de otros fármacos sobre el efavirenz) y en la tabla 3 (efecto del efavirenz sobre otros fármacos). Puede consultarse más información acerca de las recomendaciones clínicas en el apartado Interacciones medicamentosas. Tabla 2. Interacciones medicamentosas: Cambios en los parámetros farmacocinéticos del efavirenz en presencia del fármaco administrado concomitantemente.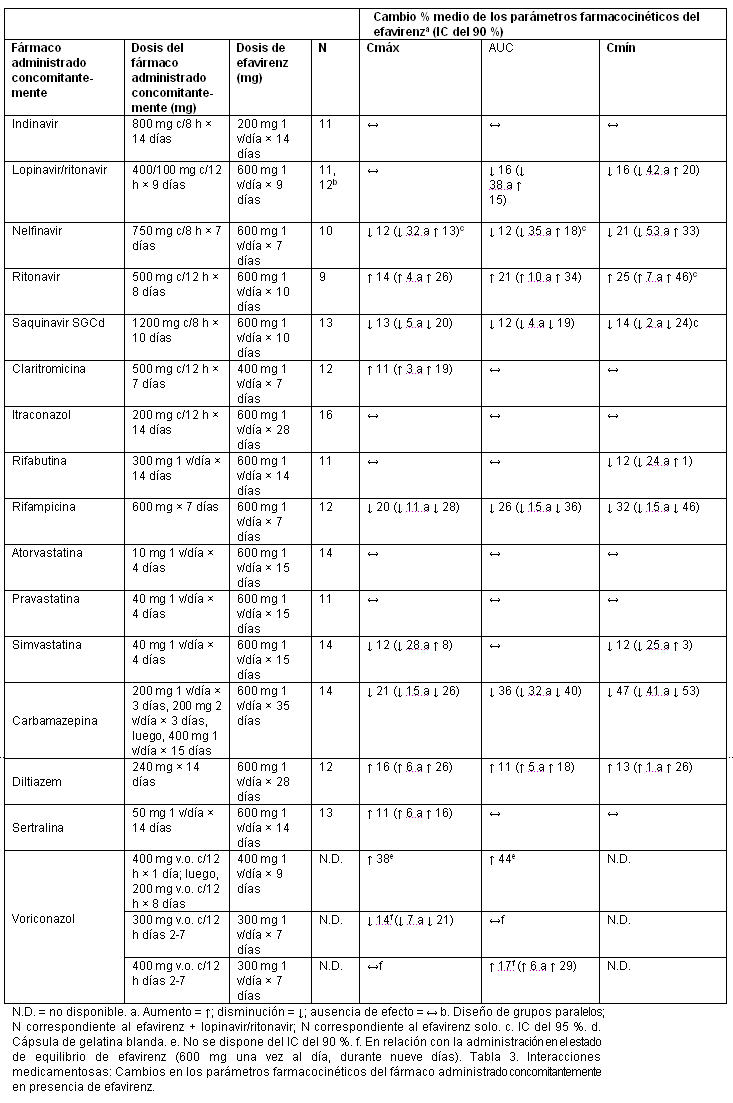
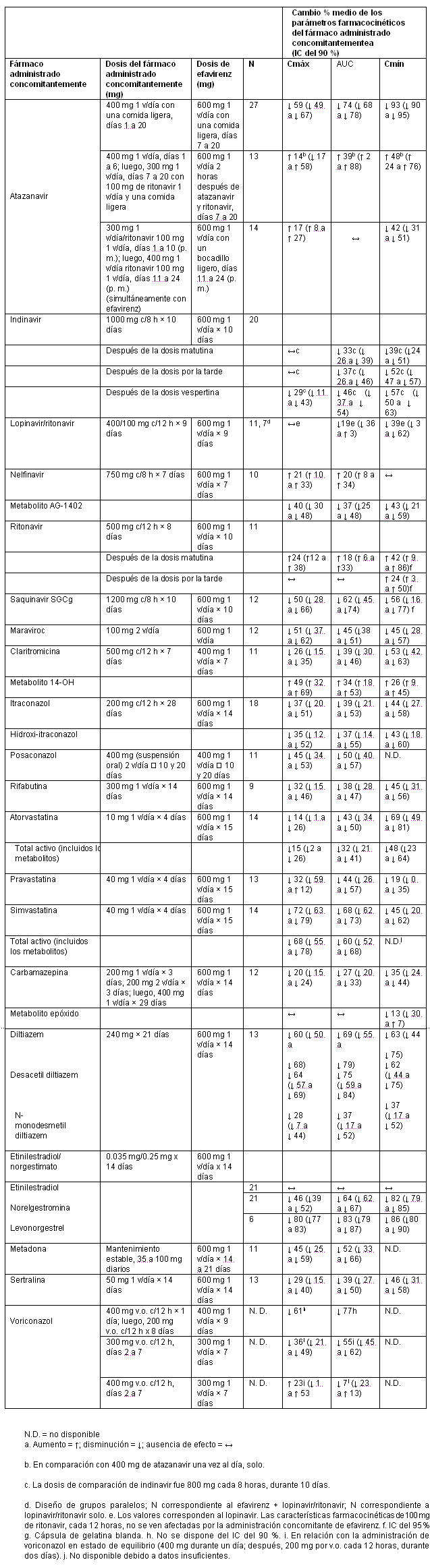
Emtricitabina y tenofovir disoproxil fumarato: Las características farmacocinéticas de la emtricitabina y del tenofovir en el estado de equilibrio no se vieron afectadas cuando se administraron emtricitabina y tenofovir disoproxil fumarato juntos, en comparación con la administración de cada fármaco por separado. En los estudios de interacción medicamentosa in vitro y en las propiedades farmacocinéticas clínicas se ha demostrado que la posibilidad de interacciones mediadas por el CYP que afectan a la emtricitabina y al tenofovir con otros medicamentos es baja. La emtricitabina y el tenofovir se excretan principalmente por los riñones, mediante una combinación de filtración glomerular y secreción tubular activa. No se observaron interacciones medicamentosas debido a la competencia por la excreción renal. Sin embargo, la administración de emtricitabina y tenofovir DF concomitantemente con medicamentos que se eliminan por secreción tubular activa puede aumentar las concentraciones de emtricitabina, tenofovir o del fármaco administrado concomitantemente.Los fármacos que disminuyen la función renal también pueden aumentar las concentraciones de emtricitabina o tenofovir. No se observaron interacciones medicamentosas clínicamente significativas entre la emtricitabina y el famciclovir, el indinavir, la estavudina, el tenofovir disoproxil fumarato y la zidovudina. De manera parecida, en ensayos realizados en voluntarios sanos, no se observaron interacciones medicamentosas clínicamente significativas entre el tenofovir disoproxil fumarato y el abacavir, el efavirenz, la emtricitabina, el entecavir, el indinavir, la lamivudina, la asociación de lopinavir y ritonavir, la metadona, el nelfinavir, los anticonceptivos orales, la ribavirina, la asociación de saquinavir y ritonavir, o el tacrolimus. Después de administrar dosis múltiples a pacientes negativos para el VIH que estaban recibiendo tratamiento de mantenimiento crónico con metadona, anticonceptivos orales o dosis únicas de ribavirina, las características farmacocinéticas del tenofovir en el estado de equilibrio fueron parecidas a las observadas en ensayos anteriores, lo que indica la ausencia de interacciones medicamentosas clínicamente significativas entre estos fármacos y el tenofovir DF. Los efectos de los fármacos administrados concomitantemente sobre la Cmáx, el AUC y la Cmín del tenofovir se muestran en la tabla 4. Los efectos de la administración concomitante de tenofovir DF sobre la Cmáx, el AUC y la Cmín de los fármacos administrados concomitantemente se muestran en las tablas 5 y 6.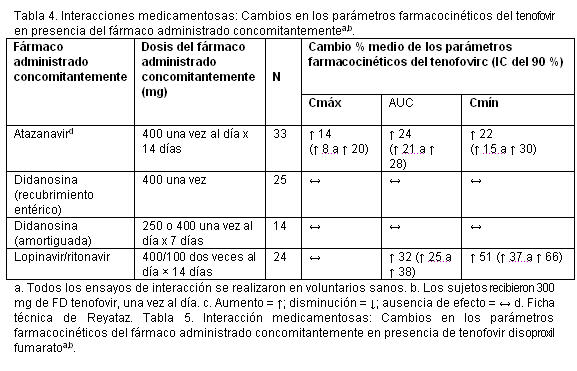
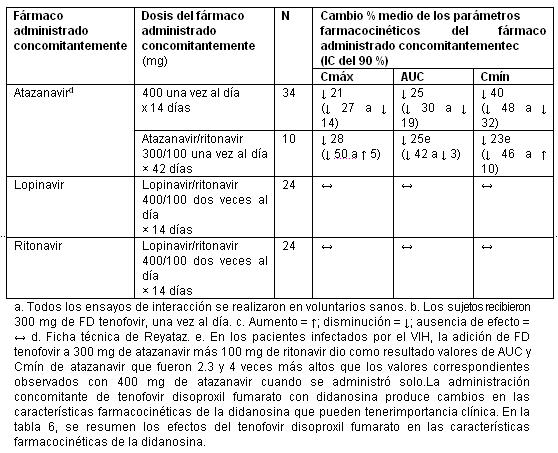
La administración concomitante de tenofovir disoproxil fumarato con didanosina produce cambios en las características farmacocinéticas de la didanosina que pueden tenerimportancia clínica. En la tabla 6, se resumen los efectos del tenofovir disoproxil fumarato en las características farmacocinéticas de la didanosina. La administración concomitante de tenofovir disoproxil fumarato con tabletas amortiguados o con cápsulas con recubrimiento entérico de didanosina aumenta significativamente la Cmáx y el AUC de la didanosina. Cuando se administraron cápsulas con recubrimiento entérico de 250 mg de didanosina con tenofovir disoproxil fumarato, las exposiciones sistémicas a la didanosina fueron parecidas a las observadas con las cápsulas con recubrimiento entérico de 400 mg solas, administradas en ayunas. Se desconoce el mecanismo de esta interacción [véanse las recomendaciones de ajuste de la posología de la didanosina en Interacciones medicamentosas, tabla 9].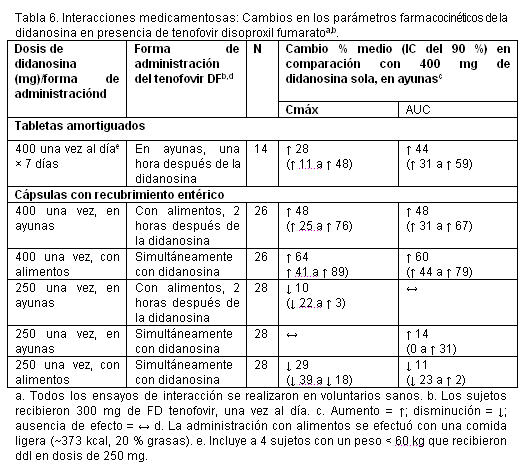
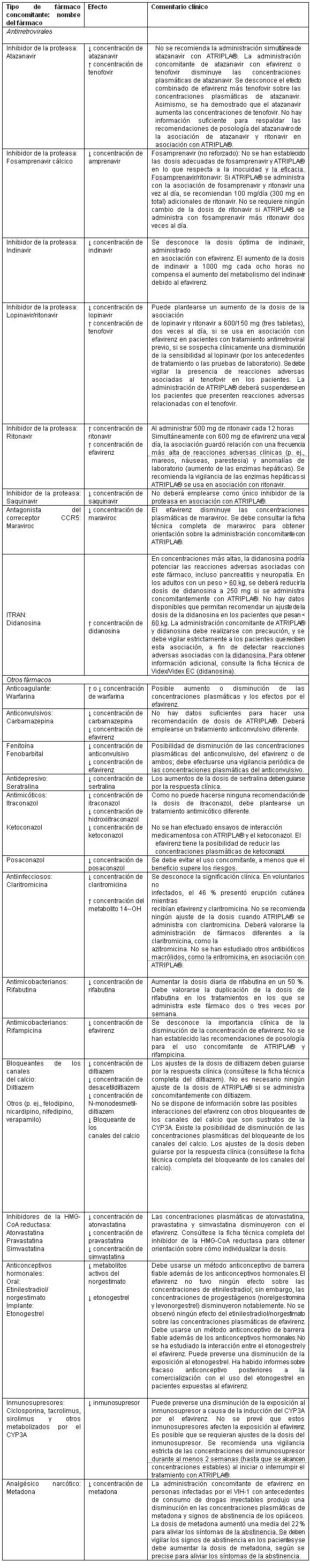
Contraindicaciones: ATRIPLA® está contraindicado en los pacientes con hipersensibilidad clínicamente significativa demostrada previamente (p. ej., Síndrome de Stevens-Johnson, eritema multiforme o erupciones cutáneas tóxicas) al efavirenz, un componente de ATRIPLA®.Fármacos contraindicados: En el caso de algunos fármacos, la competencia por el CYP3A por parte del efavirenz podría causar la inhibición de su metabolismo y crear la posibilidad de reacciones adversas graves o que pueden poner en peligro la vida del paciente (por ejemplo, arritmias cardiacas, sedación prolongada o depresión respiratoria). En la tabla 7, se enumeran los fármacos que están contraindicados con ATRIPLA®.Tabla 7. Fármacos cuyo uso está contraindicado o no se recomienda con ATRIPLA®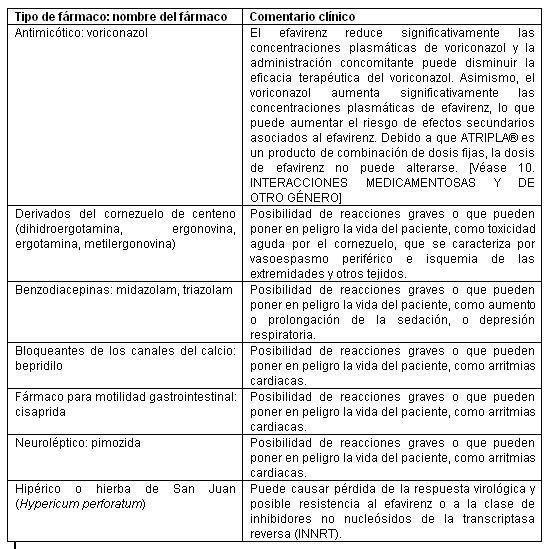
Precauciones generales: Acidosis láctica y hepatomegalia grave con esteatosis Se ha notificado la aparición de acidosis láctica y hepatomegalia grave con esteatosis, incluso casos mortales, con el uso de los análogos nucleósidos, incluido el tenofovir DF, un componente de ATRIPLA®, en combinación con otros antirretrovirales. La mayoría de estos casos se registraron en mujeres. La obesidad y la exposición prolongada a los nucleósidos pueden ser factores de riesgo. Se debe tener especial precaución cuando se administran análogos nucleósidos a cualquier paciente con factores de riesgo conocidos para las enfermedades hepáticas. Sin embargo, también se han notificado casos en los pacientes que no tenían factores de riesgo conocidos. El tratamiento con ATRIPLA® se deberá interrumpir en cualquier paciente que presente resultados clínicos o de laboratorio que sugieran acidosis láct