AVASTIN®
ROCHE
Denominación genérica: Bevacizumab.
Forma farmacéutica y formulación: Solución inyectable. El frasco ámpula contiene: bevacizumab 100 mg. Vehículo cbp 4 ml. Bevacizumab 400 mg. Vehículo cbp 16 ml.
Indicaciones terapéuticas: Cáncer colorrectal metastásico (CCRm). AVASTIN®, en combinación con quimioterapia estándar basada en fluoropirimidinas, está indicado para el tratamiento de pacientes con carcinoma metastásico de colon o recto. Cáncer de mama localmente recurrente o metastásico (CMm). AVASTIN®, en combinación con paclitaxel, está indicado para el tratamiento de primera línea de pacientes con cáncer de mama localmente recurrente o metastásico. Cáncer pulmonar de células no pequeñas (CPCNP), localmente avanzado, metastásico o recurrente. AVASTIN®, además de la quimioterapia basada en platino, está indicado para el tratamiento de primera línea de pacientes con cáncer pulmonar de células no pequeñas, no escamosas, localmente avanzado, inoperable, metastásico o recurrente, diferente al de histología celular predominantemente escamosa. Cáncer de células renales avanzado y/o metastásico. AVASTIN®, en combinación con el interferón alfa-2a, está indicado como tratamiento de primera línea de pacientes con cáncer de células renales avanzado y/o metastático.
Farmacocinética y farmacodinamia: Farmacodinamia: mecanismo de acción: AVASTIN® (bevacizumab) es un anticuerpo monoclonal recombinante humanizado que se une selectivamente y neutraliza la actividad biológica del factor de crecimiento endotelial vascular humano (VEGF). Bevacizumab contiene regiones estructuradas humanas con regiones de enlace a antígenos de un anticuerpo murino humanizado que se une a VEGF. Bevacizumab es producido por tecnología recombinante del DNA en un sistema de expresión de células de mamífero en el ovario del hámster chino, en un medio nutritivo que contiene al antibiótico gentamicina y es purificado por un proceso que incluye la inactivación viral y pasos de retiro específicos. La gentamicina es detectable en el producto final, en una cantidad de ≤0,35 ppm. Bevacizumab está integrado por 214 aminoácidos y tiene un peso molecular de aproximadamente 149.000 daltons. AVASTIN® inhibe la unión del VEGF, a sus receptores, Flt-1 y KDR, sobre la superficie de las células endoteliales. Al neutralizar la actividad biológica del VEGF se reduce la vascularización de los tumores, inhibiendo por lo tanto el crecimiento tumoral. La administración de bevacizumab o de su anticuerpo murino relacionado a modelos de xenotransplante de cáncer en ratones no manipulados, produjo una extensa actividad antitumoral en cánceres humanos que incluyeron al de colon, mama, páncreas y próstata. La progresión de la enfermedad metastásica fue inhibida y disminuyó la permeabilidad microvascular. Eficacia clínica: cáncer colorrectal metastásico (CCRm): la seguridad y eficacia de la dosis recomendada de AVASTIN® (5 mg/kg de peso corporal cada dos semanas) en carcinoma metastásico de colon o recto fue estudiada en tres estudios clínicos controlados con activo, en combinación con quimioterapia de primera línea basada en fluoropirimidinas. AVASTIN® fue combinado con dos esquemas de quimioterapia: AVF2107g: un esquema semanal de irinotecan/bolo de 5 fluorouracilo/leucovorin (esquema IFL) durante un total de 4 semanas de cada ciclo de 6 semanas. AVF0780g: en combinación con bolo de 5 fluorouracilo/leucovorin (5 FU/LV) durante un total de 6 semanas de cada ciclo de 8 semanas (esquema de Roswell Park). AVF2192g: en combinación con bolo de 5-fluorouracilo/leucovorin (5-FU/LV) durante un total de 6 semanas de cada ciclo de 8 semanas (esquema de Roswell Park) en pacientes que no eran candidatos óptimos para tratamiento de primera línea con irinotecan. Se condujeron dos estudios adicionales en primera (NO16966) y segunda línea (E3200) de tratamiento de carcinoma metastásico de colon o recto, con AVASTIN® administrado en los siguientes regímenes de dosis, en combinación con FOLFOX-4 (5FU/LV/oxaliplatino) y XELOX (capecitabina/oxaliplatino): NO16966: AVASTIN® 7,5 mg/kg de peso corporal cada 3 semanas en combinación con capecitabina oral y oxaliplatino intravenoso (XELOX) o AVASTIN® 5 mg/kg cada 2 semanas en combinación con leucovorin más bolo de 5-fluorouracilo, seguido por infusión de 5-florouracilo, con oxaliplatino intravenoso (FOLFOX-4). E3200: AVASTIN® 10 mg/kg de peso corporal cada 2 semanas en combinación con leucovorin y 5-fluorouracilo en bolo, seguido por infusión de 5-fluorouracilo, con oxaliplatino intravenoso (FOLFOX-4). AVF2107g: este fue un estudio clínico aleatorizado de fase III, doble ciego, controlado con activo, para evaluar a AVASTIN® en combinación con IFL como tratamiento de primera línea para el carcinoma metastásico de colon o recto. Ochocientos trece pacientes fueron asignados aleatoriamente para recibir IFL+ placebo (Brazo 1) o IFL+AVASTIN® (5 mg/kg cada 2 semanas, Brazo 2). Un tercer grupo de 110 pacientes recibió bolo de 5 FU/LV+AVASTIN® (Brazo 3). El reclutamiento de pacientes en el Brazo 3 fue suspendido, como estaba previamente especificado, una vez que se estableció la seguridad de AVASTIN® con el esquema de IFL y fue considerado como aceptable. El parámetro primario de eficacia del estudio, fue la supervivencia global. La adición de AVASTIN® a IFL produjo un incremento estadísticamente significativo en la supervivencia global, en la supervivencia libre de progresión y en la tasa de respuesta global (vea la tabla 1). El beneficio clínico de AVASTIN®, medido por la supervivencia, fue observado en todos los subgrupos preespecificados de pacientes, que incluyeron a aquellos definidos por la edad, sexo, estado de desempeño físico, localización del tumor primario, número de órganos afectados y duración de la enfermedad metastásica.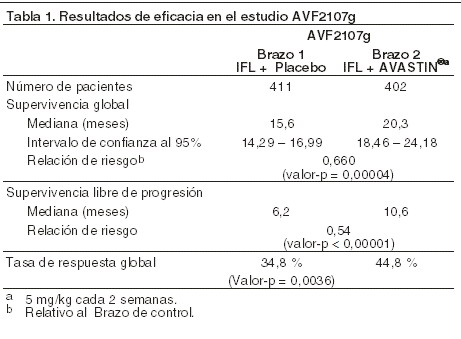
De los 110 pacientes asignados aleatoriamente al Brazo 3 (5-FU/LV + AVASTIN®), previo a la discontinuación la mediana de la supervivencia global fue de 18,3 meses, la mediana del tiempo de supervivencia libre de progresión fue de 8,8 meses. AVF2192g: éste fue un ensayo clínico, fase II, aleatorizado, doble ciego, controlado con activo que investigó AVASTIN® en combinación con 5-FU/leucovorin como tratamiento de primera línea para el cáncer colorrectal metastásico, en pacientes que no eran candidatos óptimos para el tratamiento de primera línea con irinotecan. Ciento cinco pacientes fueron aleatorizados al brazo de 5 FU/LV placebo y 104 pacientes, aleatorizados a 5-FU/LV + AVASTIN® (5 mg/kg cada 2 semanas). Todos los tratamientos continuaron hasta la progresión de la enfermedad. La adición de AVASTIN® 5 mg/kg cada dos semanas a 5-FU/LV resultó en tasas de respuesta objetiva más altas, en supervivencia libre de progresión significativamente más larga y en una tendencia de supervivencia más larga, en comparación con la quimioterapia de 5-FU/LV sola. NO16966: éste fue un ensayo clínico fase III, aleatorizado, doble ciego (para bevacizumab), que investigó AVASTIN® 7,5 mg/kg en combinación con capecitabina oral y el oxaliplatino IV (XELOX), administrado en un calendario de 3 semanas; o AVASTIN® 5 mg/kg en combinación con leucovorin con 5-fluorouracilo en bolo, seguido por 5-fluorouracilo en infusión, con oxaliplatino IV (FOLFOX-4), administrado en un calendario quincenal. El estudio comprendió dos partes: una parte inicial no ciega de 2 brazos (parte I) en la que los pacientes fueron aleatorizados a dos diferentes grupos de tratamiento (XELOX y FOLFOX-4) y una parte posterior de 4 brazos, de diseño factorial 2 x 2 (parte II) en la que los pacientes fueron aleatorizados a cuatro grupos de tratamiento (XELOX + placebo, FOLFOX-4 + placebo, XELOX +AVASTIN®, FOLFOX-4 + AVASTIN®). En la parte II, la asignación del tratamiento fue doble ciega en cuanto a AVASTIN®. Aproximadamente 350 pacientes fueron aleatorizados en cada uno de los 4 brazos del estudio en la parte II del ensayo.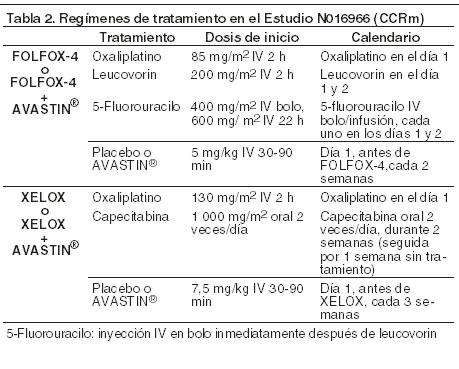
El parámetro primario de eficacia del ensayo fue la duración de la supervivencia libre de progresión. En este estudio, hubo dos objetivos primarios: mostrar que XELOX no era inferior a FOLFOX-4 y mostrar que AVASTIN® en combinación con FOLFOX-4 o con la quimioterapia XELOX era superior a la quimioterapia sola. Se cumplieron ambos objetivos primarios: 1) No se demostró inferioridad de los brazos que contenían XELOX comparados con los brazos que contenían FOLFOX-4 en la comparación global en términos de supervivencia libre de progresión (SLP) y en la supervivencia global en la población de protocolo elegible. 2) Se demostró la superioridad de los brazos que contenían AVASTIN® contra los brazos de quimioterapia sola en la comparación global, en términos de supervivencia libre de progresión en la población de intención de tratamiento ITT (tabla 3). Los análisis secundarios de la supervivencia libre de progresión, basados en el análisis del Comité de Revisión Independiente (CRI) con base en las evaluaciones de la respuesta, confirmaron el beneficio clínico superior para los pacientes tratados con AVASTIN® (los análisis de subgrupo se muestran en la tabla 3), consistente con el beneficio estadísticamente significativo observado en el análisis del acumulado.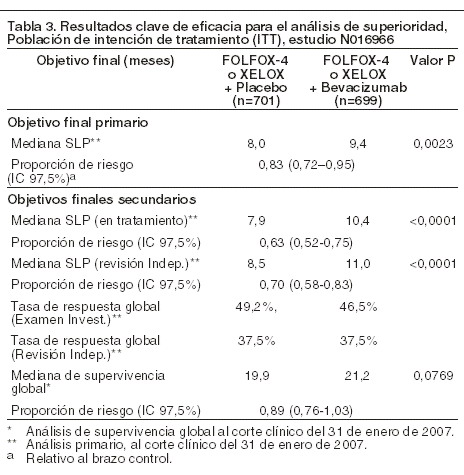
ECOG E3200: éste fue un estudio fase III, aleatorizado, controlado con activo, abierto que investigó AVASTIN® 10 mg/kg en combinación con leucovorin con 5-fluorouracilo en bolo y después 5-fluorouracilo en infusión, con oxaliplatino IV (FOLFOX-4), administrado en un calendario quincenal en pacientes previamente tratados (segunda línea) con cáncer colorrectal avanzado. En los brazos de quimioterapia, el régimen FOLFOX-4 usó las mismas dosis y calendario como se muestra en la tabla 2 del estudio NO16966. El parámetro primario de eficacia del ensayo fue la supervivencia global, definida como el tiempo desde la aleatorización hasta la muerte por cualquier causa. Ochocientos veintinueve pacientes fueron aleatorizados (292 a FOLFOX-4, 293 a AVASTIN® + FOLFOX-4 y 244 a la monoterapia de AVASTIN®). La adición de AVASTIN® a FOLFOX-4 resultó en una prolongación estadísticamente significativa de la supervivencia. También se observaron mejorías estadísticamente significativas en la supervivencia libre de progresión y en la tasa de respuesta objetiva (ver la tabla 4).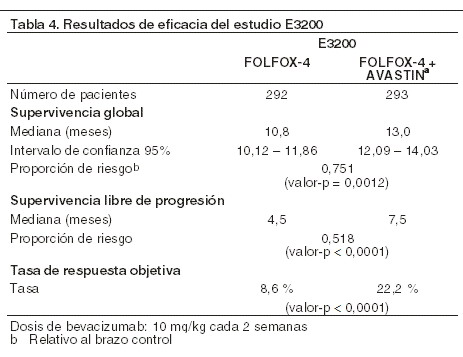
No se observó diferencia significativa en la duración de la supervivencia global entre los pacientes que recibieron la monoterapia de AVASTIN® comparada con los pacientes tratados con FOLFOX-4. La supervivencia libre de progresión y la tasa de respuesta objetiva fueron inferiores en el brazo de monoterapia de AVASTIN® en comparación con el brazo de FOLFOX-4. Cáncer de mama localmente recurrente o metastásico (CMm): el E2100 fue un estudio clínico abierto, aleatorizado, con control activo, multicéntrico que evaluó AVASTIN® en combinación con paclitaxel para cáncer de mama localmente recurrente o metastásico en pacientes que no habían recibido quimioterapia previa para enfermedad localmente recurrente y metastásica. Se permitió la terapia hormonal previa para el tratamiento de la enfermedad metastásica. Se permitía la terapia adyuvante con taxanos sólo si se completaba al menos 12 meses antes del ingreso al estudio. Las pacientes fueron aleatorizadas a paclitaxel solo (90 mg/m2 IV durante una hora una vez a la semana por tres de cuatro semanas) o en combinación con AVASTIN® (10 mg/kg en infusión IV cada dos semanas). Los pacientes iban a continuar siendo asignados al tratamiento del estudio hasta la progresión de la enfermedad. En los casos en los que los pacientes suspendieran la quimioterapia de forma prematura, se continuaba el tratamiento con AVASTIN® como agente único hasta la progresión de la enfermedad. El parámetro primario fue la supervivencia libre de progresión (SLP), evaluado por el investigador. Adicionalmente, se condujo una revisión independiente del objetivo primario: de los 722 pacientes en el estudio, la mayoría de los pacientes (90%) tuvieron enfermedad HER-2 negativa. Una pequeña cantidad de pacientes tuvo estado del receptor HER-2 que se desconocía (8%) o era positivo (2%). Los pacientes que fueron positivos a HER-2 habían recibido tratamiento previo con trastuzumab o eran considerados inapropiados para trastuzumab. La mayoría (65%) de los pacientes habían recibido quimioterapia adyuvante incluyendo el 19% que había recibido taxanos con anterioridad y el 49% que había recibido antraciclinas anteriormente. Las características del paciente fueron similares entre los brazos del estudio. Los resultados de este estudio se presentan en la tabla 5.
Cáncer pulmonar de células no pequeñas (CPCNP), localmente avanzado metastásico o recurrente: se estudió la seguridad y eficacia de AVASTIN® en el tratamiento de primera línea de pacientes con cáncer pulmonar de células no pequeñas (CPCNP) diferente al de histología de células predominantemente escamosas, además de la quimioterapia con base en platino en los estudios E4599 y BO17704. El estudio E4599 fue abierto, aleatorizado, controlado activamente y multicéntrico para evaluar el tratamiento de primera línea con AVASTIN® de pacientes con CPCNP localmente avanzado, metastático o recurrente con histología diferente a las células predominantemente escamosas. Los pacientes se aleatorizaron en grupos con quimioterapia basada en platino (paclitaxel 200 mg/m2 y carboplatino ABC= 6,0, ambos por infusión IV) (PC) en el día 1 de cada ciclo de tres semanas hasta 6 ciclos de PC solo o en combinación con AVASTIN® a una dosis de 15 mg/kg de infusión IV en el día 1 de cada ciclo de 3 semanas. Después de completar 6 ciclos de quimioterapia carboplatino-paclitaxel o hasta la discontinuación prematura de la quimioterapia, los pacientes en el brazo de AVASTIN®+ carboplatino-paclitaxel continuaron recibiendo AVASTIN® como agente único cada 3 semanas hasta la progresión de la enfermedad. Se aleatorizaron 878 pacientes en los dos brazos. Durante el estudio, de los pacientes que recibieron el tratamiento del estudio, el 32,2% (136/422) de los pacientes recibieron de 7-12 tomas de AVASTIN® y el 21,1% (89/422) de los pacientes recibieron 13 o más dosis de AVASTIN®. El objetivo final primario fue la duración de la supervivencia. Los resultados se presentan en la tabla 6.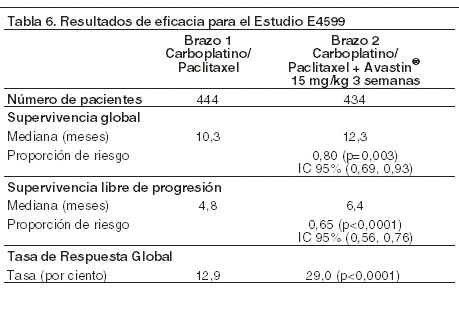
El estudio BO17704 es un estudio actualmente activo fase III, aleatorizado, doble ciego, de AVASTIN® en adición a cisplatino y gemcitabina contra placebo, cisplatino y gemcitabina en pacientes con CPCNP no escamoso, localmente avanzado, metastático o recurrente diferente del de histología celular predominantemente escamosa, que no haya recibido quimioterapia anteriormente. El objetivo final primario es la supervivencia libre de progresión (SLP). Los pacientes se aleatorizaron a la quimioterapia basada en platino, infusión IV de cisplatino 80 mg/m2 el día 1 e infusión IV de gemcitabina 1250 mg/m2 en los días 1 y 8 de cada ciclo de tres semanas hasta por 6 ciclos (CG) con placebo o CG en combinación con una infusión IV de AVASTIN® a una dosis de 7,5 o 15 mg/kg el día 1 de cada ciclo de tres semanas. En los brazos que contienen AVASTIN®, después de completar la quimioterapia, se permitió que los pacientes recibieran AVASTIN® como agente único cada 3 semanas hasta la progresión de la enfermedad o toxicidad no aceptable. Los resultados del estudio mostraron que 94% (277/298) de los pacientes elegibles recibieron el agente único bevacizumab en el ciclo 7. Los resultados parciales de eficacia se presentan en la tabla 7.
Cáncer de células renales avanzado y/o metastásico: BO17705 fue un ensayo multicéntrico, aleatorizado, doble ciego, fase III conducido para evaluar la eficacia y la seguridad de AVASTIN® en combinación con interferón (IFN)-alfa-2a contra IFN-alfa-2a solo, como tratamiento de primera línea en el cáncer de células renales avanzado y/o metastásico de células claras. Los 649 pacientes aleatorizados (641 tratados) tuvieron estado de desempeño Karnofsky (KPS) ≥70%, sin metástasis al SNC y función adecuada de órganos. IFN-alfa-2a (x3/semana a la dosis recomendada de 9 MIU) más AVASTIN® (10mg/kg cada 2 semanas) o placebo fue administrado hasta la progresión de la enfermedad. Los pacientes fueron estratificados de acuerdo al país y a la puntuación de Motzer, y se mostró que los brazos de tratamiento estuvieron bien balanceados para los factores de pronóstico. En el corte de datos, habían ocurrido 505 eventos de progresión, 111 pacientes permanecieron con tratamiento, 287 habían suspendido (las suspensiones del tratamiento del ensayo debido a eventos adversos fueron del 12% con IFN vs. 28% con IFN-alfa-2a/AVASTIN®), y 251 murieron. Noventa y siete pacientes en el brazo de IFN alfa-2a y 131 pacientes en el brazo de AVASTIN® disminuyeron la dosis de IFN- alfa-2a de 9 MIU a 6 o 3 MIU, tres veces a la semana como se especificó en el protocolo previamente. La reducción de la dosis de IFN alfa-2a no parece afectar la eficacia de la combinación de AVASTIN® e IFN alfa-2a basada en el evento SLP exento de tasas sobre el tiempo, como se muestra en el análisis de subgrupos. Los 131 pacientes en el AVASTIN® + el brazo IFN alfa 2a en el cual la dosis de IFN alfa-2a a 6 o 3 MIU durante el estudio esta reducida y mantenida, muestran a los 6, 12 y 18 meses del evento SLP exento de tasas del 73, 52 y 21% respectivamente, al compararlo con el 61, 43 y 17% en la población total de pacientes que recibieron AVASTIN® + IFN alfa-2a. La adición de AVASTIN® a IFN-alfa-2a aumentó significativamente la supervivencia libre de progresión (SLP) y la tasa de respuesta objetiva del tumor (vea la tabla 8). Los datos de la supervivencia global (SG) no estaban concluidos al momento del análisis preliminar programado. Se observó una tendencia hacia la mejoría en la SG con la adición de AVASTIN® a IFN-alfa-2a (p=0,0670).
AVF2938: éste fue un estudio clínico aleatorizado, doble ciego, fase II que investigó AVASTIN® 10mg/kg en un calendario quincenal con la misma dosis de AVASTIN® en combinación con 150 mg diarios de erlotinib, en pacientes con cáncer de células renales de estirpe células claras. Se aleatorizó un total de 104 pacientes al tratamiento en este estudio, 53 a AVASTIN® 10 mg/kg, cada 2 semanas más placebo y 51 a AVASTIN® 10 mg/kg cada 2 semanas más erlotinib 150 mg diario. El análisis del parámetro primario no mostró diferencia entre el brazo de AVASTIN® + placebo y el brazo de AVASTIN® + erlotinib (mediana de la SLP 8,5 contra 9,9 meses). Siete pacientes en cada brazo tuvieron una respuesta objetiva. Farmacocinética: la farmacocinética de bevacizumab se caracterizó en los pacientes con varios tipos de tumores sólidos. Las dosis probadas fueron de 0,1-10 mg/kg a la semana, en la fase I; de 3-20 mg/kg cada 2 semanas (c2s) o cada tres semanas (c3s) en la fase II; de 5 mg/kg (c2s) o 15 mg/kg c3s en la fase III. En todos los ensayos, se administró bevacizumab como infusión IV. Como se observó con otros anticuerpos, la farmacocinética de bevacizumab está bien descrita por un modelo de dos compartimentos. En general, en todos los ensayos clínicos, la disposición de bevacizumab se caracterizó por una baja depuración, un volumen limitado del compartimiento central (Vc), y una vida media de larga eliminación. Esto permite que se mantengan los niveles plasmáticos, terapéuticos de bevacizumab dentro del rango de los períodos de administración (como una administración cada 2 o 3 semanas). En el análisis de la población de farmacocinética, no hubo diferencia significativa en la farmacocinética de bevacizumab en relación con la edad (sin correlación entre la depuración de bevacizumab y la edad del sujeto [la mediana de la edad fue de 59 años con percentiles 5° y 95° de 37 y 76 años]). Los niveles bajos de albúmina y elevados de fosfatasa alcalina generalmente son indicativos de la severidad de la enfermedad y la carga tumoral. La depuración de bevacizumab fue aproximadamente 20% más elevada, ya sea en los sujetos con niveles bajos de albúmina en suero o en los sujetos con niveles elevados de fosfatasa alcalina, cuando se compararon con un sujeto típico con la mediana de valores de albúmina y/o fosfatasa alcalina. Absorción: no aplicable. Distribución: el valor típico para Vc fue de 2,66 l y de 3,25 l para mujeres y hombres respectivamente, que es el rango que se ha descrito para las IgGs y otros anticuerpos monoclonales. Después de corregir con el peso corporal, los sujetos hombres tuvieron un Vc mayor (+ 22%) que las mujeres. Metabolismo: la evaluación del metabolismo de bevacizumab en conejos, después de administrar una dosis única IV de 125I-bevacizumab, indicó que su perfil metabólico fue similar al esperado para la molécula nativa de IgG, la cual no se une al VEGF. El metabolismo y la eliminación de bevacizumab es similar al IgG endógeno, por ej., la vía proteolítica principal del catabolismo a través del cuerpo, incluyendo las células endoteliales, no dependiendo primariamente de la eliminación a través de riñones y el hígado. El enlace de IgG al receptor FcRn da como resultado en la protección del metabolismo celular y el tiempo de vida media terminal prolongada. Eliminación: la farmacocinética de bevacizumab es lineal a las dosis que oscilan entre 1,5 y 10 mg/kg/semana. La depuración de bevacizumab fue de un promedio de 0,207 L/día para las mujeres y de 0,262 l/día en los hombres. Después de realizar la corrección del peso corporal, los hombres tuvieron una mayor depuración de bevacizumab (26%) que las mujeres. De acuerdo con el modelo bicompartimental, la vida media inicial (a) es de 1,4 días para ambos sexos, y el estimado de la vida media (b) terminal es de 20 días para una mujer típica y de 19 días para un hombre típico. Farmacocinética en poblaciones especiales: la farmacocinética de la población se analizó para evaluar los efectos de las características demográficas. Los resultados no mostraron una diferencia significativa en la farmacocinética de bevacizumab en relación con la edad. Niños y adolescentes: la farmacocinética de bevacizumab ha sido estudiada en un número limitado de pacientes pediátricos. Los datos resultantes de la farmacocinética sugieren que el volumen de distribución y la eliminación de bevacizumab fueron comparables con aquellos adultos que tenían tumores sólidos. Daño renal: no se han llevado a cabo estudios para investigar la farmacocinética del bevacizumab en pacientes con daño renal desde que los riñones no son los órganos mejor conocidos para el metabolismo o eliminación. Daño hepático: no se han realizado estudios para la investigación de la farmacocinética de bevacizumab en pacientes con fallas hepáticas puesto que el hígado no es el órgano mejor conocido para el metabolismo o eliminación del bevacizumab. Seguridad preclínica displasia fiscal: en estudios hasta de 26 semanas de duración en monos Cynomolgus, AVASTIN® fue asociado a displasia fiseal. La displasia fiseal estuvo caracterizada principalmente por engrosamiento de los cartílagos de crecimiento, formación de placas de hueso subcondral e inhibición de las placas de crecimiento. Este efecto ocurrió en dosis ≥0,8 veces la dosis terapéutica recomendada para el humano y con niveles de exposición ligeramente menores a los esperados en el humano, en base al promedio de las concentraciones séricas. Debe hacerse notar, sin embargo, que la displasia fiseal ocurrió únicamente en animales en fase de crecimiento activo, con placas de crecimiento abiertas. Debido a que AVASTIN® será administrado con mayor probabilidad a pacientes adultos con placas de crecimiento cerradas, no es de esperarse que se presente displasia fiseal en la población clínica. Cicatrización de heridas: se estudiaron los efectos de AVASTIN® sobre la cicatrización de las heridas circulares en los conejos. La reepitelización de la herida fue retrasada en los conejos, después de la administración de cinco dosis de AVASTIN® en dosis que se encontraron en el rango de 2 a 50 mg/kg, durante un período de 2 semanas. Se observó una tendencia hacia una relación dependiente de la dosis. La magnitud del efecto sobre la cicatrización de la herida fue similar a la observada con la administración de corticoesteroides. Al terminar el tratamiento, ya fuese con 2 o 10 mg/kg de AVASTIN®, las heridas cerraron completamente. La dosis más baja de 2 mg/kg fue aproximadamente equivalente a la dosis clínica propuesta. En los conejos también se estudió un modelo más sensible de cicatrización lineal de la herida. Tres dosis de AVASTIN® en el rango de 0,5 a 2 mg/kg dosis-dependiente, disminuyeron significativamente la fuerza tensional de las heridas, consistentemente con un retraso de la cicatrización. La dosis baja de 0,5 mg/kg fue 5 veces más baja de la dosis clínica propuesta. Debido a que los efectos sobre la cicatrización de la herida fueron observados en los conejos, en dosis por debajo de la dosis clínica propuesta, deberá considerarse la capacidad de AVASTIN® para afectar adversamente la cicatrización de heridas en el humano. En los monos Cynomolgus (macaco de Java), los efectos de AVASTIN® sobre la cicatrización de una incisión lineal, fueron altamente variables y no hubo evidencia de una relación dosis-respuesta. Función renal: en los monos Cynomolgus normales, tratados una o dos veces por semana, hasta por 26 semanas, AVASTIN® no tuvo un efecto medible sobre la función renal y no se acumuló en el riñón de los conejos, después de dos dosis de hasta 100 mg/kg (aproximadamente 80 veces la dosis clínica propuesta). Los estudios de toxicidad realizados en los conejos, utilizando los modelos de disfunción renal, mostraron que AVASTIN® no exacerbó la lesión glomerular renal inducida por la albúmina sérica de bovino ni el daño tubular renal inducido por cisplatino. Albúmina: en los monos Cynomolgus machos (macaco de Java), AVASTIN® administrado en dosis de 10 mg/kg, dos veces por semana o 50 mg/kg una vez a la semana, durante 26 semanas, se asoció con una disminución estadísticamente significativa en la relación de la albúmina, albúmina/globulina y un incremento en la globulina. Esos efectos fueron reversibles al terminar la exposición. Debido a que los parámetros permanecieron dentro del rango de referencia normal, de los valores para esos parámetros, esos cambios no fueron considerados como clínicamente significativos. Hipertensión: en las dosis de hasta 50 mg/kg dos veces a la semana, en monos Cynomolgus, AVASTIN® no mostró efectos sobre la presión arterial. Hemostasis: los estudios de toxicología preclínica de hasta 26 semanas de duración, realizados en monos Cynomolgus, no se encontraron cambios en la biometría hemática ni en los parámetros de coagulación, que incluyeron las cuentas de plaquetas, tiempo de protrombina y tiempo de tromboplastina parcial activado. Un modelo de hemostasis en conejos, utilizado para investigar el efecto de AVASTIN® sobre la formación de trombos, no mostró ninguna alteración sobre la tasa de formación de coágulos ni en ningún otro parámetro hematológico, en comparación al tratamiento con el vehículo AVASTIN®.
Contraindicaciones: AVASTIN® está contraindicado en los pacientes con hipersensibilidad conocida a: cualquiera de los componentes del producto. A los productos de células de ovario de hámster chino o a otros anticuerpos humanos recombinantes o humanizados. AVASTIN® está contraindicado en los pacientes con metástasis no tratadas en el sistema nervioso central (SNC) (ver Precauciones generales y Reacciones secundarias y adversas).
Precauciones generales: Perforaciones gastrointestinales: los pacientes pueden estar en riesgo incrementado de desarrollar perforación gastrointestinal, cuando son tratados con AVASTIN® (ver Reacciones secundarias y adversas). AVASTIN® deberá suspenderse permanentemente en los pacientes que desarrollen perforación gastrointestinal. Fístulas: los pacientes pueden tener un riesgo incrementado para desarrollar fístulas cuando son tratados con AVASTIN®. Discontinuar permanentemente AVASTIN® en los pacientes con fístula traqueoesofágica (TE) o cualquier fístula de grado 4. Está disponible información limitada sobre el uso continuo de AVASTIN® en pacientes con otras fístulas. En los casos de fístulas internas que surgen en sitios diferentes al tracto GI, se deberá considerar la discontinuación de AVASTIN®. Hemorragia (ver Reacciones secundarias y adversas): los pacientes tratados con AVASTIN® tienen un mayor riesgo de presentar hemorragias, especialmente las asociadas a tumores. AVASTIN® deberá suspenderse permanentemente en los pacientes que experimenten sangrado de grado 3 o 4 durante la terapia con AVASTIN®. El riesgo de hemorragia en el SNC en pacientes con metástasis en el SNC que están recibiendo AVASTIN® no pudo ser evaluado completamente, debido a que estos pacientes fueron excluidos de los estudios clínicos. No existe información sobre el perfil de seguridad de AVASTIN® en pacientes con diátesis hemorrágica congénita, coagulopatía adquirida o en los pacientes que reciben dosis completas de anticoagulantes para el tratamiento de tromboembolismo previo al inicio del tratamiento con AVASTIN®, debido a que dichos pacientes fueron excluidos de los estudios clínicos. Por lo tanto, se debe tener precaución antes de iniciar la terapia con AVASTIN® en esos pacientes. Sin embargo, los pacientes que desarrollaron trombosis venosa mientras estaban recibiendo la terapia con AVASTIN® no mostraron un incremento en la tasa de sangrado grado 3 o superior grave cuando fueron tratados concomitantemente con dosis completas de warfarina y AVASTIN®. Hemorragia pulmonar/hemoptisis: los pacientes con cáncer pulmonar de células no pequeñas tratados con AVASTIN® pueden estar bajo riesgo de hemorragia pulmonar/hemoptisis grave y en algunos casos fatal (ver Reacciones secundarias y adversas. Hemorragia). Los pacientes con hemorragia/hemoptisis pulmonar reciente ( > ½ cucharadita de sangre) no deberán tratarse con AVASTIN®. Hipertensión: se observó un incremento en la incidencia de hipertensión en los pacientes tratados con AVASTIN®. Los datos de seguridad clínica sugieren que la incidencia de hipertensión probablemente sea dependiente de la dosis. La hipertensión preexistente se debe controlar adecuadamente antes de iniciar el tratamiento con AVASTIN®. No existe información sobre el efecto de AVASTIN® en los pacientes con hipertensión no controlada al momento de iniciar con la terapia de AVASTIN®. Se recomienda el monitoreo de la presión arterial durante la terapia con AVASTIN® (ver Reacciones secundarias y adversas). En la mayoría de los casos, la hipertensión fue controlada adecuadamente usando tratamiento antihipertensivo estándar adecuado para la situación individual del paciente afectado. AVASTIN® se debe suspender de forma permanente si la hipertensión médicamente significativa no se puede controlar adecuadamente con terapia antihipertensiva, o si el paciente desarrolla crisis hipertensiva o encefalopatía hipertensiva (ver Reacciones secundaria y adversas y postcomercialización). Síndrome de leucoencefalopatía posterior reversible (SLPR): ha habido raros reportes de pacientes tratados con AVASTIN® que desarrollaron signos y síntomas que sean consistentes con el síndrome de leucoencefalopatía posterior reversible (SLPR), trastorno neurológico raro que puede presentarse con los siguientes signos y síntomas entre otros: convulsiones, cefalea, estado mental alterado, alteración visual o ceguera cortical, con o sin hipertensión asociada. El diagnóstico de SLPR requiere confirmación mediante imagen cerebral. En los pacientes que desarrollan SLPR, se recomienda el tratamiento de los síntomas específicos, incluyendo el control de la hipertensión, junto con la suspensión de AVASTIN®. Se desconoce la seguridad del reinicio de la terapia con AVASTIN® en los pacientes que experimentaron previamente SLPR (ver postcomercialización). Tromboembolismo arterial (ver Reacciones secundarias): en estudios clínicos, la incidencia de eventos de tromboembolismo arterial, incluyendo los eventos vasculares cerebrales, las crisis isquémicas transitorias (CIT) y los infartos del miocardio (IM), fue más elevada en pacientes que estaban recibiendo AVASTIN® en combinación con quimioterapia, en comparación con aquellos quienes recibieron la quimioterapia sola. AVASTIN® deberá suspenderse permanentemente en los pacientes que desarrollen eventos tromboembólicos arteriales. Pacientes que recibieron AVASTIN® más quimioterapia con antecedentes de tromboembolismo arterial o una edad mayor de 65 años, se asociaron con un incremento en el riesgo de eventos tromboembólicos arteriales, durante la terapia con AVASTIN®. Se debe tener precaución al tratar a estos pacientes con AVASTIN®. Tromboembolismo venoso: los pacientes pueden estar en riesgo de desarrollar eventos tromboembólicos venosos, incluyendo embolismo pulmonar bajo tratamiento con AVASTIN®. AVASTIN® se debe suspender en los pacientes con embolismo pulmonar (grado 4) que ponga en riesgo la vida, en pacientes con ≤ grado 3 que necesiten ser controlados de forma estrecha. Insuficiencia cardíaca congestiva: en los ensayos clínicos se reportaron eventos consistentes con insuficiencia cardíaca congestiva (ICC). Los síntomas variaron desde asintomático, que disminuye la fracción de eyección ventricular izquierda, hasta la ICC sintomática, que requirieron tratamiento u hospitalización. La mayoría de los pacientes que experimentaron ICC tenían cáncer de mama metastásico y habían recibido tratamiento previo con antraciclinas, radioterapia previa en la pared izquierda del tórax u otros factores de riesgo para ICC, tales como enfermedad cardíaca coronaria preexistente o terapia cardiotóxica concomitante. Se debe tener precaución al tratar con AVASTIN® a pacientes con enfermedad cardiovascular clínicamente significativa o insuficiencia cardíaca congestiva preexistente. Neutropenia: se han observado tasas elevadas de neutropenia severa, neutropenia febril o infección con neutropenia severa (incluyendo algunas víctimas mortales) en los pacientes tratados con algún régimen de quimioterapia mielotóxica más AVASTIN®, en comparación con la quimioterapia sola. Cicatrización de heridas: AVASTIN® puede afectar en forma adversa el proceso de cicatrización de las heridas. La terapia con AVASTIN® no deberá iniciarse durante por lo menos 28 días después de una cirugía mayor o hasta que la herida quirúrgica haya cicatrizado completamente. En los pacientes que experimenten complicaciones en la cicatrización de las heridas durante el tratamiento con AVASTIN®, el medicamento deberá suspenderse hasta que la herida haya cicatrizado completamente. La terapia con AVASTIN® deberá suspenderse en caso de una cirugía electiva (ver Reacciones secundarias y adversas). Proteinuria (ver Reacciones secundarias): en los ensayos clínicos, la incidencia de proteinuria fue más alta en los pacientes que recibieron AVASTIN® en combinación con quimioterapia en comparación con aquéllos que recibieron quimioterapia sola. La proteinuria grado 4 (síndrome nefrótico) no fue común en los pacientes con AVASTIN®. En caso de proteinuria grado 4, se debe suspender AVASTIN® de forma permanente. Efectos sobre la capacidad para conducir automóvil y el uso de maquinaría: no se han realizado estudios acerca de los efectos sobre la capacidad de conducir un automóvil y el uso de maquinaria. Sin embargo, no existe evidencia de que el tratamiento con AVASTIN® resulta en un incremento de eventos adversos que podrían conducir a una alteración en la capacidad para conducir un automóvil u operar maquinaria o una alteración en la capacidad mental. Uso pediátrico: no se ha estudiado la seguridad y la eficacia de AVASTIN® en niños y adolescentes. Uso geriátrico: referirse a la sección Tromboembolismo Arterial. Deterioro renal: no se ha estudiado la seguridad y la eficacia de AVASTIN® en pacientes con deterioro renal. Deterioro hepático: no se ha estudiado la seguridad y la eficacia de AVASTIN® en pacientes con deterioro hepático.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: la angiogénesis ha demostrado ser de importancia crítica para el desarrollo fetal. La inhibición de la angiogénesis tras la administración de AVASTIN® podría traducirse en un resultado adverso del embarazo. No existen estudios adecuados y bien controlados en mujeres embarazadas (ver Teratogénesis). Se sabe que las IgG cruzan la barrera placentaria y AVASTIN® puede inhibir la angiogénesis en el feto. Por lo tanto, AVASTIN® no deberá ser utilizado durante el embarazo. En las mujeres con potencial de concebir, se recomiendan medidas anticonceptivas apropiadas durante la terapia con AVASTIN®. Con base en consideraciones de farmacocinética, se recomiendan medidas anticonceptivas por lo menos durante 6 meses después de la última dosis de AVASTIN®. Lactancia: se desconoce si bevacizumab es excretado en la leche humana. Debido a que la IgG es excretada en la leche y AVASTIN® podría dañar el crecimiento y desarrollo del infante, a las madres se les debe aconsejar que no amamanten a sus bebés durante la terapia con AVASTIN® y no reiniciar la alimentación al pecho durante por lo menos 6 meses después de la administración de la última dosis de AVASTIN®.
Reacciones secundarias y adversas: Experiencia obtenida de los estudios clínicos: se han conducido ensayos clínicos en más de 3.500 pacientes con varias neoplasias malignas, predominantemente tratadas con AVASTIN®, principalmente en combinación con quimioterapia. El perfil de seguridad de la población del ensayo clínico