BARACLUDE®
BRISTOL M.S.
Denominación genérica: Entecavir.
Forma farmaceutica y formulación: Tabletas. Cada tableta contiene: Entecavir 0.5 mg y 1 mg. Excipiente cbp 1 tableta.
Descripción: BARACLUDE® (entecavir) es un análogo de nucleósido de guanosina con actividad potente y selectiva contra el virus de la hepatitis B (VHB). BARACLUDE® está disponible para su administración oral como tabletas recubiertas de 0.5 mg y 1 mg de entecavir.
Indicaciones terapéuticas: BARACLUDE® está indicado para el tratamiento de la infección crónica por VHB en adultos con evidencia de inflamación activa del hígado.
Farmacocinética y farmacodinamia en humanos: Microbiología: Mecanismo de acción: Entecavir es un análogo de nucleósido de guanosina con una actividad potente y selectiva contra la polimerasa del VHB. Es fosforilado a la forma activa trifosfato (TP), que tiene una vida media intracelular de 15 horas. Los niveles intracelulares de TP se relacionan directamente con las concentraciones extracelulares de entecavir, sin acumulaciones importantes más allá de los niveles de meseta. Al competir con el sustrato desoxiguanosina- TP natural, entecavir-TP inhibe las 3 actividades funcionales de la polimerasa viral: (1) inicio de actividad de la polimerasa del VHB, (2) la transcripción inversa de la cadena negativa del ARN mensajero pregenómico, y (3) la síntesis de la cadena positiva del ADN del VHB. El entecavir-TP Ki para la polimerasa del ADN del VHB es 1.2 nM. El entecavir-TP es un inhibidor débil de las polimerasas a, b y d del ADN celular con valores para Ki de 18 a 40 mM. Además, las altas exposiciones al entecavir-TP y el entecavir no tuvieron efectos adversos relevantes sobre la polimerasa c (Ki > 160 mM) o la síntesis del ADN mitocondrial en las células HepG2. Actividad antiviral: El entecavir inhibió la síntesis del ADN del VHB (reducción del 50%, EC50) a una concentración de 0.004 mM en células HepG2 humanas transfectadas con VHB de tipo salvaje. El valor medio de EC50 para el entecavir contra el VHB resistente a la lamivudina (rtM204V, rtL180M) fue de 0.026 mM (rango 0.010-0.059 mM). Un análisis completo de la actividad inhibidora del entecavir contra un panel de aislados de VIH-1 de laboratorio y clínicos, con una variedad de células y condiciones, dio valores de EC50 s de 0.026 a > 10 mM. La actividad inhibidora a concentraciones por debajo de ~1 mM sólo se observó al modificar las condiciones de los cultivos celulares de una manera que se disminuyera el nivel del virus utilizado para iniciar la infección. En un cultivo celular, entecavir seleccionó para una sustitución M184I a concentraciones micromolares, confirmando la presión inhibitoria a altas concentraciones de entecavir. Las variantes de VIH con la sustitución M184V mostraron pérdida de susceptibilidad a entecavir. Resistencia en cultivo celular: En estudios celulares, en comparación con la actividad contra el VHB de tipo salvaje, hay una reducción de 8 veces en la susceptibilidad a entecavir cuando se encuentran presentes las sustituciones rtM204I/V ± rtL180M (LVDr) que se asocian con resistencia a lamivudina y telbivudina. A concentraciones extracelulares representativas de los niveles plasmáticos alcanzados con una dosis de 1 mg, se esperaría que los niveles intracelulares de entecavir-TP sobrepasaran los niveles necesarios para inhibir la actividad enzimática de las polimerasas del VHB resistente a la lamivudina. Los virus recombinantes que codifican las sustituciones resistentes al adefovir en rtN236T o rtA181V siguieron siendo totalmente susceptibles a entecavir. Resistencia clínica: Los pacientes en estudios clínicos inicialmente tratados con 0.5 mg de entecavir (sin tratamiento previo con nucleósidos) o 1.0 mg (resistentes a la lamivudina) y con una determinación del ADN del VHB por PCR durante tratamiento durante o después de la Semana 24 estuvieron bajo monitoreo para detectar resistencia. Las recaídas virológicas debidas a resistencia a entecavir requieren de la existencia previa de sustituciones primarias de LVDr (M204I/V ± L180M), además de una sustitución adicional en los residuos T184, S202, y/o M250 de la proteína de la polimerasa. Estudios en pacientes sin tratamiento previo con nucleósidos: Se observó evidencia genotípica de sustituciones de resistencia al entecavir (ETVr) en rtT184, rtS202, o rtM250 en 3 pacientes tratados con entecavir, 2 de los cuales experimentaron hasta por 240 semanas en estudios de pacientes sin tratamiento previo con nucleósidos. Seiscientos sesenta y tres pacientes fueron tratados y vigilados en cuanto a resistencia en el Año 1, 278 pacientes en el Año 2, 149 pacientes en el Año 3, 121 pacientes en el Año 4 y 108 pacientes en el año 5. Los resultados reflejan el uso de una dosis de 1 mg de entecavir para 147 pacientes en el Año 3, y para todos los pacientes en el Año 4 y 5, y de la terapia de combinación de entecavir-lamivudina (seguida por una terapia a largo plazo con entecavir) durante una mediana de 20 semanas para 130 pacientes en el Año 3 y durante 1 semana para 1 paciente en el Año 4 en un estudio de continuación. La probabilidad acumulada de sustituciones de ETVr genotípica en estudios de pacientes sin tratamiento previo con nucleósidos fue de 0.2%, 0.5%, 1.2%, 1.2% y 1.2 % durante el Año 1, Año 2, Año 3, Año 4 y Año 5, respectivamente. La probabilidad acumulada de recaídas virológicas con sustituciones de ETVr fue de 0.2%, 0.2%, 0.8%, 0.8% y 0.8% durante el Año 1, Año 2, Año 3, Año 4 y Año 5, respectivamente. Estudios de resistencia a la lamivudina: Los análisis genotípicos de muestras clínicas obtenidas de pacientes con resistencia a lamivudina identificaron sustituciones emergentes de ETVr en 11 de 187 de los pacientes en el Año 1, 12 de 146 de los pacientes en el Año 2, 16 de 80 de los pacientes en el Año 3; 6 de 52 de los pacientes en el Año 4 y 2 de 33 pacientes en el Año 5. En los Años 1, 2, 3, 4 y 5, 2/187, 14/146, 13/80, 9/53 y 1/33 pacientes, respectivamente, experimentaron recaída virológica (incremento ≥1 log10 por encima del nadir) con evidencia de resistencia genotípica. La probabilidad acumulada de sustituciones genotípicas emergentes de ETVr genotípica en estudios de resistencia a la lamivudina fue de 6%, 15%, 36%, 47% y 51% durante el Año 1, Año 2, Año 3, Año 4 y Año 5, respectivamente. La probabilidad acumulada de recaídas virológicas debido a ETVr fue de 1%, 11%, 27%, 41% y 44% durante el Año 1, Año 2, Año 3, Año 4 y Año 5, respectivamente. Los resultados reflejan el uso de una terapia combinada de entecavir-lamivudina (seguida por una terapia a largo plazo con entecavir) durante una mediana de 13 semanas para 48 de 80 pacientes en el Año 3 y durante una mediana de 38 semanas para 10 pacientes en el Año 4 y por 16 semanas para 1 paciente en el Año 5 respectivamente. Los resultados reflejaron que el uso en combinación de la terapia entecavir-lamivudina (seguido de una terapia de largo plazo de entecavir) por una mediana de de 13 semanas por 48 pacientes en el año 3, por una mediana de 38 semanas de 10 pacientes en el año 4 y por 16 semanas para un paciente en el Año 5, en un estudio de continuación. La presencia de sustituciones de ETVr en el basal en aislados para 10 (5%) de 187 pacientes resistentes a la lamivudina indica que el tratamiento previo con lamivudina puede seleccionar estas sustituciones de resistencia y que éstas pueden existir con una baja frecuencia antes del tratamiento con entecavir. Hasta la Semana 192, 3 de estos 10 pacientes experimentaron una recaída virológica. Farmacocinética: absorción: En sujetos sanos, entecavir se absorbió rápidamente y las concentraciones plasmáticas pico se presentaron entre 0.5 y 1.5 horas. Hubo un incremento en la concentración plasmática pico (C máx) proporcional a la dosis y los valores del área bajo la curva (ABC) tiempo-concentración después de múltiples dosis fueron de 0.1 a 1 mg. El estado estable se alcanzó después de 6-10 días de dosis una vez al día, con una acumulación de aproximadamente el doble. Los valores para Cmáx y concentración plasmática mínima (Cmín) en el estado estable fueron de 4.2 y 0.3 ng/mL, respectivamente, para una dosis de 0.5 mg, y de 8.2 y 0.5 ng/mL, respectivamente, para una dosis de 1 mg. En sujetos sanos, la biodisponibilidad de la tableta fue de 100% en relación con la solución oral. La solución oral y la tableta se pueden emplear en forma intercambiable. La administración oral de 0.5 mg de entecavir con una comida estándar alta en grasas (945 kcal, 54.6 g de grasas) o una comida ligera (379 kcal, 8.2 g de grasas) tuvo como resultado un retraso mínimo en la absorción (1-1.5 horas después de la comida vs. 0.75 horas en ayunas), una disminución en Cmáx de 44-46%, así como una disminución en la ABC de 18-20% (Ver Dosis y vía de administración). Distribución: El volumen calculado de distribución de entecavir excedió el agua corporal total, lo que sugiere que tiene una buena penetración en los tejidos. La unión a proteínas séricas en humanos in vitro fue de aproximadamente 13%. Metabolismo y eliminación: Entecavir no es un substrato, inhibidor, o inductor del sistema enzimático CYP450. A concentraciones aproximadamente 10,000 veces mayores a las obtenidas en humanos, el entecavir no inhibió ninguna de las enzimas principales 1A2, 2C9, 2C19, 2D6, 3A4, 2B6 y 2E1 del CYP450 humano. A concentraciones aproximadamente 340 veces mayores que las observadas en humanos, el entecavir no induce las enzimas 1A2, 2C9, 2C19, 3A4, 3A5 y 2B6 del CYP450 humano. Después de la administración de 14C-entecavir en humanos y ratas, no se observaron metabolitos oxidativos o acetilados ni cantidades menores de metabolitos de fase II (conjugados de glucurónido y de sulfato). Después de alcanzar niveles pico, las concentraciones plasmáticas de entecavir disminuyeron de manera biexponencial con una vida media de depuración terminal de aproximadamente 128-149 horas. El índice observado de acumulación del medicamento es aproximadamente el doble con dosis una vez al día, lo que sugiere un vida media efectiva de acumulación de aproximadamente 24 horas. Entecavir se elimina predominantemente por el riñón, con una recuperación urinaria del medicamento sin cambios en estado estable de entre 62% y 73% de la dosis. La depuración renal es independiente de la dosis y va de 360 a 471 mL/min, lo que sugiere que el entecavir pasa tanto por filtración glomerular como por secreción tubular neta. Poblaciones especiales: Pacientes con insuficiencia renal: Con base en los resultados del estudio de la farmacocinética de una sola dosis de 1 mg de entecavir en pacientes con disfunción renal (Tabla 1), se recomienda ajustar la dosificación para pacientes con una depuración de creatinina < 50 mL/min (Ver Pacientes con insuficiencia renal).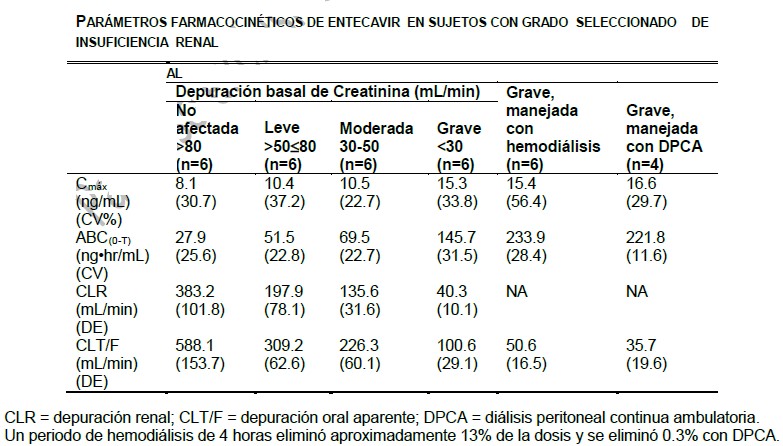
Pacientes con insuficiencia hepática: Los parámetros farmacocinéticos de entecavir en pacientes con insuficiencia hepática fueron similares a los de los pacientes con función hepática normal. Geriátricos: El perfil farmacocinético del entecavir no difiere por edad. Género/raza: El perfil farmacocinético del entecavir no difiere por género o raza (Ver Grupos raciales/étnicos). Receptores de trasplante de hígado: La exposición a entecavir en receptores de trasplante de hígado infectados por el VHB, bajo dosis estables de ciclosporina A (n=5) o tacrolimus (n=4), fue de aproximadamente el doble de la exposición en sujetos sanos, con función renal normal. La función renal alterada contribuyó al incremento en la exposición a entecavir en estos pacientes (Ver Receptores de trasplante de hígado). Información de estudios clínicos: La demostración de los beneficios de BARACLUDE® se basa en las respuestas histológicas, virológicas, bioquímicas y serológicas en pacientes adultos no tratados previamente con nucleósidos y en pacientes con resistencia a lamivudina, con infección crónica por VHB HBeAg-positivos o HBeAg-negativos y enfermedad hepática compensada. Se evaluaron la eficacia y seguridad en tres estudios clínicos controlados con tratamiento activo, que incluyeron a 1633 pacientes con infección crónica por VHB (positivos para HBsAg en suero por al menos 6 meses) acompañada por evidencia de replicación viral (ADN del VHB detectable en suero). Los pacientes presentaban niveles persistentemente elevados de ALT ≥ 1.3 veces el LSN e inflamación crónica en la biopsia del hígado, compatible con el diagnóstico de hepatitis viral crónica. De conformidad con los criterios obligatorios del protocolo, los pacientes que participaron en estos tres estudios clínicos suspendieron el tratamiento con el fármaco del estudio después de 52 semanas, de acuerdo con una definición de respuesta basada en la supresión virológica de VHB ( < 0.7 MEq/mL por ADNb) y la pérdida de HBeAg (en pacientes HBeAg-positivos) o ALT < 1.25 X LSN (en pacientes HBeAg-negativos) en la Semana 48. Los pacientes que lograron supresión virológica sin una respuesta serológica (HBeAg-positivos), o que no alcanzaron niveles de ALT < 1.25 X LSN (HBeAg-negativos), siguieron recibiendo dosificación ciega durante 98 semanas o hasta cubrir los criterios de respuesta. Estas directrices especificadas en el protocolo sobre el tratamiento de pacientes no pretenden ser una guía en la práctica clínica. También se ha estudiado BARACLUDE® en pacientes adultos infectados con VHB con enfermedad hepática compensada y en pacientes adultos coinfectados por VIH/VHB que han recibido tratamiento previo con lamivudina. Pacientes sin tratamiento previo con nucleósidos con enfermedad hepática compensada: Resultados a las 48 semanas: La Tabla 2 presenta los resultados a las 48 semanas para dos estudios aleatorizados, doble ciego, en pacientes sin tratamiento previo con nucleósidos, uno en pacientes HBeAg-positivo y otro en pacientes HBeAg- negativo, comparando entecavir con lamivudina.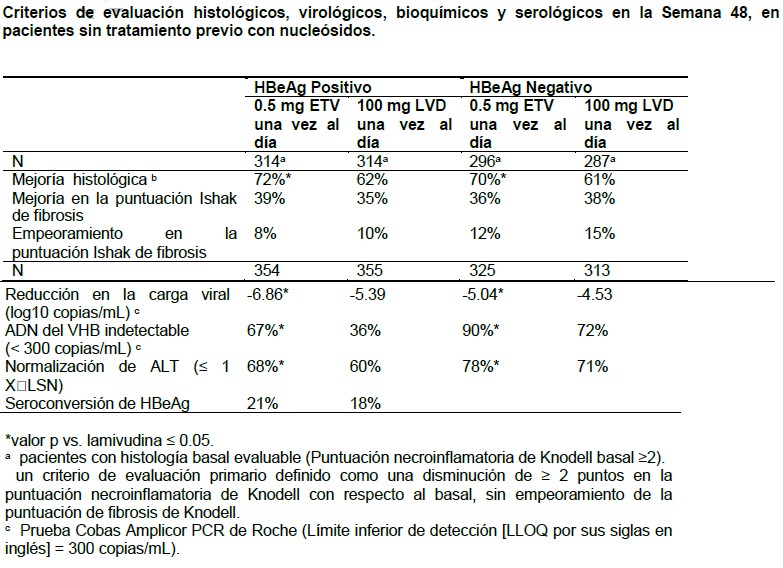
Resultados después de 48 semanas, en pacientes sin tratamiento previo con nucleósidos: HbeAg-positivos: Al final de la dosificación, entre los pacientes HBeAg-positivos que continuaron con el tratamiento después de las 52 semanas (mediana de 96 semanas), 74% de los 243 pacientes tratados con BARACLUDE® y 37% de los 164 pacientes tratados con lamivudina tenían < 300 copias/mL del ADN del VHB por PCR, mientras que la normalización de ALT (≤ 1 vez el LSN) se presentó en 79% de los pacientes tratados con BARACLUDE® y 68% de los pacientes tratados con lamivudina. Hasta las 96 semanas, los resultados confirmados acumulados para los pacientes HBeAg-positivos (todos tratados) demostraron que el tratamiento continuo con BARACLUDE® (n=354) tuvo como resultado un incremento en la proporción de pacientes con ADN del VHB < 300 copias/mL mediante la prueba PCR (80%) y normalización de ALT (87%). Hasta la última observación con o sin tratamiento, 31% de los pacientes tratados con BARACLUDE® tenían seroconversión de HBeAg y 5% tenían pérdida de HBsAg. En el grupo de tratamiento con lamivudina (n=355), el ADN acumulado confirmado del VHB era < 300 copias/mL según PCR para 39% de los pacientes y había normalización de ALT en 79%; 26% de los pacientes tenían seroconversión de HBeAg y 3% pérdida de HBsAg. La diferencia entre los grupos de tratamiento fue estadísticamente significativa para el porcentaje de pacientes con ADN del VHB < 300 copias/mL y normalización de ALT (p < 0.01). HbeAg-negativos: Para 26 pacientes HBeAg-negativos tratados con BARACLUDE® y 28 pacientes tratados con lamivudina que continuaron el tratamiento después de las 52 semanas (mediana de 96 semanas), 85% de los pacientes tratados con BARACLUDE® y 57% de los pacientes tratados con lamivudina tenían ADN del VHB < 300 copias/mL por PCR al final de la dosificación. La normalización de ALT (≤ 1 vez el LSN) se presentó en 27% de los pacientes tratados con BARACLUDE® y 21% de los pacientes tratados con lamivudina al final de la dosificación. Hasta las 96 semanas, en los pacientes HBeAg-negativos, 94% de los pacientes tratados con BARACLUDE® (n=325) y 77% de los pacientes tratados con lamivudina (n=313) tenían un ADN acumulado confirmado del VHB de < 300 copias/mL (p < 0.01). La normalización de ALT se presentó en 89% de los pacientes tratados con BARACLUDE® y 84% de los pacientes tratados con lamivudina Resultados de biopsia de hígado: De 679 pacientes tratados con BARACLUDE® en dos estudios sin tratamiento previo de nucleósidos, 293 (43%) de los pacientes elegibles fueron enrolados en un estudio de repetición y continuaron la terapia con BARACLUDE®. Los pacientes en el estudio de repetición recibieron 1 mg de BARACLUDE® una vez al día. Sesenta y nueve de 293 pacientes fueron elegidos para repetir la biopsia de hígado después de un tratamiento total de más de 144 semanas de duración (3 años). Cincuenta y siete pacientes tenían tanto una línea base evaluable y una biopsia de largo plazo con una terapia de duración mediana de BARACLUDE® de 280 semanas (aproximadamente 6 años). Noventa y seis por ciento de estos pacientes tuvieron una mejoría Histológica como se definió previamente (ver tabla 2, pie de página b) y 88% tuvieron una disminución ≥1 punto en la puntuación de fibrosis de Ishak. De los 43 pacientes con una calificación en la línea base en la fibrosis de Ishak de ≥2, el 58% tuvieron una disminución ≥2 puntos. En el momento de la biopsia, 57 (100%) de pacientes tuvieron HBV DNA < 300 copias/ml y 49 (86%) tuvieron suero ALT < 1 X LSN. Experiencia en pacientes resistentes a la lamivudina: Resultados a las 48 semanas: La Tabla 3 presenta los resultados a las 48 semanas de un estudio aleatorizado, doble ciego, que comparó el entecavir con la lamivudina en pacientes HBeAg-positivos resistentes a la lamivudina, donde 85% de los pacientes presentaban mutaciones de LVDr en la determinación basal.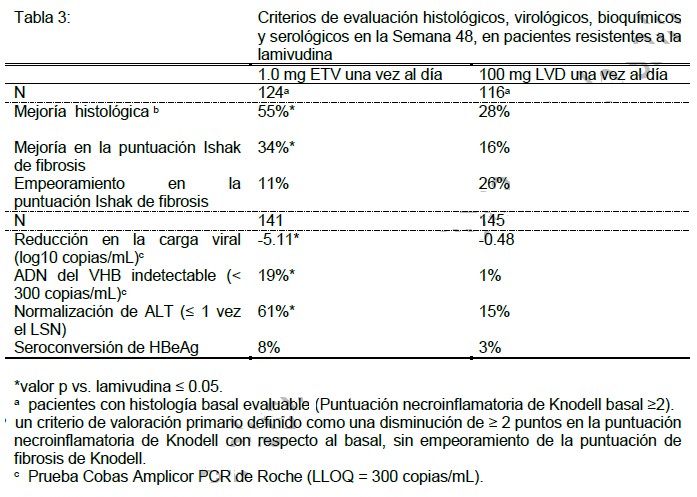
Resultados después de 48 semanas: Para los 77 pacientes resistentes a lamivudina que continuaron con el tratamiento con BARACLUDE® después de 52 semanas (mediana de 96 semanas), 40% de los pacientes tenían < 300 copias/mL de ADN del VHB según PCR y 81% tenían Normalización de ALT (≤ 1 vez el LSN) al final de la dosificación. Los resultados acumulados confirmados hasta las 96 semanas para todos los pacientes tratados resistentes a la lamivudina (n=141) demuestran que el tratamiento continuo con BARACLUDE® resulta en un incremento en la proporción de pacientes con < 300 copias/mL de ADN del VHB según PCR (30%) y normalización de ALT (85%). Hasta la última observación con o sin tratamiento, 17% de los pacientes tratados con BARACLUDE® presentaban seroconversión de HBeAg. La diferencia entre los grupos tratados con BARACLUDE® y con lamivudina fue estadísticamente significativa para los tres parámetros (p < 0.01). Seguimiento posterior al tratamiento: Para el 31% de los pacientes sin tratamiento previo con nucleósidos, HBeAg-positivos tratados con BARACLUDE®, que cumplieron con los criterios de respuesta (supresión virológica por prueba de ADNb y pérdida de HBeAg) y que suspendieron el tratamiento, la respuesta fue sostenida en 75% durante las 24 semanas de seguimiento posterior al tratamiento. Para 88% de los pacientes sin tratamiento previo con nucleósidos, HBeAg-negativos tratados con BARACLUDE®, que cumplieron con los criterios de respuesta (supresión virológica por prueba de ADNb y ALT < 1.25 X LSN), la respuesta fue sostenida en 46% durante las 24 semanas de seguimiento posterior al tratamiento. De los 22 (16%) pacientes resistentes a la lamivudina que cumplieron con los criterios de respuesta (supresión virológica por prueba de ADNb y pérdida de HBeAg) bajo administración de BARACLUDE®, la respuesta fue sostenida en 11 (50%) durante las 24 semanas de seguimiento posterior al tratamiento. Poblaciones especiales: Pacientes con enfermedad hepática descompensada: En un estudio abierto aleatorizado, 191 pacientes con infección crónica por VHB HBeAg-positivo o negativo y con evidencia de descompensación hepática, definida como una escala Child-Turcotte-Pugh (CTP) de 7 o más, recibieron 1 mg de BARACLUDE® una vez al día o adefovir dipivoxil 10 mg una vez al día. Se incluyeron pacientes sin experiencia previa al tratamiento contra VHB o pretratados (excluyendo los pretratamientos con BARACLUDE®, adefovir dipivoxil, o tenofovir disoproxil fumarato). BARACLUDE® fue superior a adefovir dipivoxil en el desenlace primario de cambio promedio del valor basal en DNA de VHB en suero mediante PCR en 24 semanas. Los resultados de algunos desenlaces a las 24 y 48 semanas son mostrados en la tabla 4: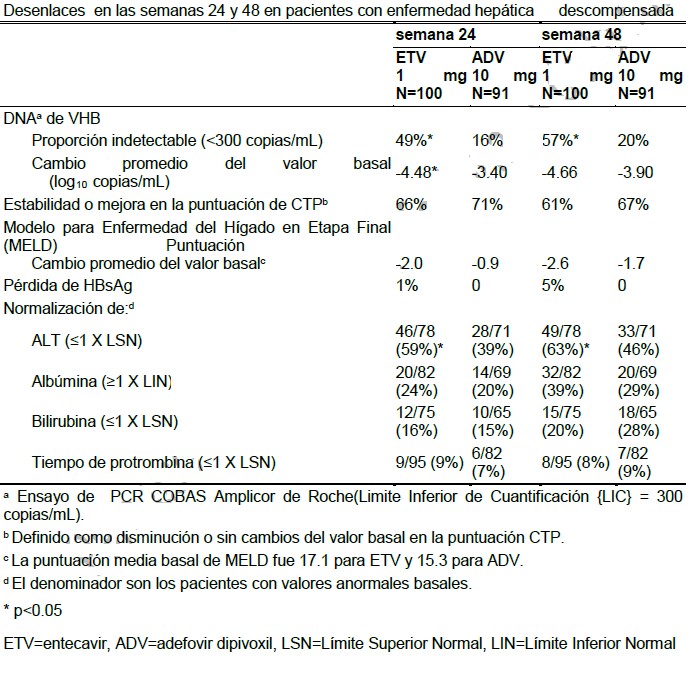
El tiempo de presentación de Carcinoma Hepatocelular (CCC) o muerte (cualquiera que ocurriera primero) fue comparable en los dos grupos de tratamiento. Pacientes coinfectados con VIH y VHB-: Un estudio aleatorizado, doble ciego, controlado con placebo, comparó BARACLUDE® con placebo en 68 pacientes coinfectados con VIH/VHB que presentaban recurrencia de viremia por VHB en un régimen de HAART que incluía lamivudina. Los pacientes continuaron con el régimen de lamivudina y se les asignó a 1 mg de BARACLUDE® una vez al día (n=51) o placebo (n=17) durante 24 semanas, seguidas por una fase abierta durante 24 semanas más, durante las cuales todos recibieron BARACLUDE®. A las 24 semanas, se observó una reducción en la carga viral de VHB por PCR con BARACLUDE® (-3.65 log10 copias/mL), mientras que se midió un ligero incremento con el placebo (+0.11 log10 copias/mL, p < 0.0001). Seis por ciento de los pacientes tratados con BARACLUDE® y ninguno de los pacientes tratados con placebo tuvieron < 300 copias/mL de ADN del VHB por PCR, mientras que 34% de los pacientes tratados con BARACLUDE® y 8% de los pacientes tratados con placebo que tenían ALT anormal en la determinación basal tuvieron normalización de ALT (≤1 X LSN). Al final de la fase abierta (Semana 48), el cambio medio con respecto al basal en los niveles de ADN del VHB por PCR en los pacientes asignados originalmente a BARACLUDE® fue de -4.20 log10 copias/mL; 8% de los pacientes tenían < 300 copias/mL de ADN del VHB por PCR; y 37% de los pacientes con ALT anormal en el basal tuvieron normalización de ALT. No se ha evaluado BARACLUDE® en pacientes coinfectados con VIH/VHB que no están recibiendo de manera concomitante tratamiento efectivo para el VIH (ver Precauciones generales / advertencias y precauciones específicas del producto / Coinfección con VIH).
Contraindicaciones: BARACLUDE® está contraindicado para pacientes con hipersensibilidad previamente demostrada a entecavir o cualquier componente del producto.
Precauciones generales: Advertencias y precauciones específicas para la clase de medicamento: Acidosis láctica / hepatomegalia con esteatosis: Se han reportado casos de acidosis láctica y hepatomegalia severa con esteatosis, incluyendo casos fatales, con el uso de los análogos de nucleósidos solos o en combinación con antirretrovirales. Exacerbaciones de la hepatitis después de suspender el tratamiento: Se ha reportado exacerbación aguda de la hepatitis en pacientes que han suspendido la terapia para la hepatitis B, incluyendo la terapia con BARACLUDE®. (Ver Reacciones adversas - exacerbaciones de la hepatitis después de suspender el tratamiento). La mayoría de las exacerbaciones posteriores al tratamiento parecen ser auto-limitadas. Sin embargo, se pueden presentar exacerbaciones graves, incluyendo muertes. Se desconoce la relación causal de estos eventos con la suspensión de la terapia. Después de suspender la terapia, se debe supervisar la función hepática a intervalos repetidos. Si es adecuado, se debe garantizar el reinicio del tratamiento para la hepatitis B. Advertencias y precauciones específicas para el producto: Coinfección con VIH: No se ha evaluado BARACLUDE® en pacientes coinfectados con el virus de inmunodeficiencia humana (VIH) y el VHB que no estén recibiendo en forma concurrente un tratamiento efectivo contra el VIH. La experiencia clínica limitada sugiere que existe el potencial para el desarrollo de resistencia al VIH cuando se utiliza BARACLUDE® para el tratamiento crónico de la infección por hepatitis B en pacientes con infección por VIH no tratada (ver Farmacocinética y farmacodinamia en humanos / microbiología / Actividad antiviral). Por lo tanto, no se recomienda la terapia con BARACLUDE® en pacientes coinfectados con VIH/VHB que no están recibiendo terapia antirretroviral altamente activa (HAART). Ver Reacciones secundarias y adversas / Pacientes coinfectados con VIH e y Farmacocinética y farmacodinamia en humanos / Poblaciones Especiales / Pacientes coinfectados con VIH y VHB para datos de eficacia y seguridad de BARACLUDE®, de un estudio de pacientes coinfectados con VIH/VHB bajo régimen HAART con lamivudina. No se ha estudiado BARACLUDE® como tratamiento para la infección por VIH y no se recomienda para este uso. Uso en poblaciones específicas: Pacientes con insuficiencia renal: Se recomienda ajustar la dosis de BARACLUDE® para pacientes con disfunción renal (Ver Dosis y vía de administración: Pacientes con disfunción renal). Receptores de trasplante de hígado: Hay datos limitados disponibles sobre la seguridad y eficacia de BARACLUDE® en receptores de trasplante hepático. En un estudio abierto, de un solo brazo, donde los pacientes que tenían menos de un 172 UI/mL de ADN de VHB al momento del trasplante fueron tratados con BARACLUDE® 1 mg una vez al día después del trasplante. La frecuencia y naturaleza de los eventos adversos en este estudio fueron consistentes con las esperadas en pacientes que habían recibido un trasplante de hígado y el perfil de seguridad conocido de BARACLUDE®. En receptores de trasplante de hígado que reciben algún inmunosupresor que puede afectar la función renal, como ciclosporina o tacrolimus, se debe mantener vigilancia de la función renal antes y durante la terapia con BARACLUDE® (Ver Dosis y vía de administración: Pacientes con insuficiencia hepática, Farmacocinética y farmacodinamia en humanos / poblaciones especiales/ pacientes con insuficiencia hepática, y Receptores de transplante de hígado). Grupos raciales/étnicos: Los datos limitados de un estudio abierto de un solo brazo de BARACLUDE® en una población predominantemente de raza Negra/Afroamericana sin tratamiento previo con nucleósidos e infección crónica por VHB demostraron seguridad y eficacia virológica que son consistentes con las observadas en los ensayos clínicos controlados descritos en Farmacocinética y farmacodinamia en humanos / poblaciones especiales / pacientes sin tratamiento previo con nucleósidos con enfermedad hepática compensada. Información para el paciente: Los pacientes deben ser advertidos, que la terapia con Baraclude® no ha demostrado reducción del riesgo de transmisión de VHB y por lo tanto, todavía se deben tomar las precauciones adecuadas
Restricciones de uso durante el embarazo y la lactancia: No hay estudios adecuados y bien controlados en mujeres embarazadas. Sólo se debe usar BARACLUDE® en el embarazo si los beneficios potenciales justifican el riesgo potencial para el feto. No hay datos sobre el efecto del entecavir sobre la transmisión materno-infantil del VHB. Por lo tanto, se deben utilizar medidas adecuadas para evitar la adquisición neonatal del VHB. Entecavir se excreta en la leche de ratas. Se desconoce si se excreta en la leche humana. Se debe indicar a las madres que eviten la lactancia si están tomando BARACLUDE®
Reacciones secundarias y adversas: La evaluación de las reacciones adversas se basa en cuatro estudios clínicos, en donde 1720 pacientes con infección crónica por VHB y hepática compensada recibieron tratamiento doble ciego con 0.5 mg/día de BARACLUDE® (n = 679), 1 mg/día de BARACLUDE® (n = 183), o lamivudina (n = 858) hasta por 107 semanas. En estos estudios, los perfiles de seguridad de BARACLUDE® y lamivudina fueron comparables. Entre los pacientes tratados con BARACLUDE®, los eventos adversos más comunes, de cualquier gravedad, con al menos una posible relación a BARACLUDE® fueron dolor de cabeza (9%), fatiga (6%), mareos (4%) y náuseas (3%). En estos estudios clínicos, los 594 pacientes tratados con BARACLUDE® que recibieron terapia ciega durante más de 52 semanas reportaron reacciones adversas de naturaleza y severidad similares a las reportadas durante las primeras 52 semanas de tratamiento. Eventos clínicos: Pacientes sin tratamiento previo con nucleósidos: En dos estudios doble ciego, controlados con lamivudina, uno con pacientes que fueron positivos para el antígeno e de la hepatitis B (HBeAg) y uno con pacientes HBeAg-negativos, 679 pacientes sin tratamiento previo con nucleósidos recibieron 0.5 mg de BARACLUDE® una vez al día durante una mediana de 54 semanas. Las reacciones adversas de intensidad moderada o mayor que se consideran al menos posiblemente relacionadas con el tratamiento con BARACLUDE® aparecen enumeradas por órganos y sistemas. La frecuencia se define como muy común (≥ 1/10); común (≥ 1/100, < 1/10); no común (≥ 1/1,000, < 1/100). Trastornos psiquiátricos: no común: insomnio. Trastornos del sistema nervioso: común: dolor de cabeza, no común: mareos, somnolencia. Trastornos gastrointestinales: no común: náuseas, diarrea, dispepsia, vómito. Trastornos generales y condiciones del sitio de administración:
común: fatiga. Pacientes que no responden a Lamivudina: En dos estudios doble ciego, controlados con lamivudina, 183 pacientes resistentes a lamivudina recibieron 1 mg de BARACLUDE® una vez al día durante una mediana de 69 semanas. Las reacciones adversas de intensidad moderada o mayor que se consideran posiblemente relacionadas al tratamiento con BARACLUDE aparecen enumeradas por órganos y sistemas. La frecuencia se define como muy común (≥ 1/10); común (≥ 1/100, < 1/10); y no común (≥ 1/1,000, < 1/100). Trastornos del sistema nervioso: común: dolor de cabeza. Trastornos gastrointestinales: común: diarrea, dispepsia. Trastornos generales y condiciones del sitio de administración: común: fatiga. Exacerbaciones de la hepatitis después de la suspensión del tratamiento: Se han reportado exacerbaciones agudas de la hepatitis en pacientes que han suspendido la terapia anti-VHB, incluyendo la terapia con BARACLUDE®. (Ver Advertencias y precauciones). La TABLA 5 presenta la frecuencia de exacerbación de la hepatitis o la elevación de ALT (definida como una ALT > 10 X LSN y 2 X por arriba del valor de referencia del paciente), durante el seguimiento en estudios clínicos con BARACLUDE®.
Poblaciones especiales: Enfermedad hepática descompensada: Otras reacciones adversas observadas en pacientes tratados con BARACLUDE® en un estudio en el cual BARACLUDE® 1 mg/día fue comparado con dipivoxilo de adefovir en pacientes con infección crónica por hepatitis B y enfermedad hepática descompensada incluyen disminución de bicarbonato en sangre (2%) e insuficiencia renal ( < 1%). El estudio sobre la tasa de mortalidad acumulada fue de 23% (23/102), y las causas de muerte fueron generalmente relacionadas al hígado, como se esperaba en esta población. La tasa acumulada de carcinoma hepatocelular (HCC) durante el estudio fue de 12% (12/102). Pacientes coinfectados con VIH: Los pacientes coinfectados con VHB y VIH que experimentaron recurrencia de la viremia de VHB mientras recibían un régimen antirretroviral altamente activo con lamivudina recibieron tratamiento con su régimen que incluía lamivudina (300 mg/día) y 1 mg de Baraclude® una vez al día (n=51) o placebo (n=17). Después de 24 semanas de terapia doble ciego y un promedio de 17 semanas de terapia abierta (durante la cual todos los pacientes recibieron Baraclude®), los perfiles de eventos adversos y anormalidades de laboratorio fueron similares para los grupos de tratamiento con Baraclude® y placebo. No se ha evaluado Baraclude® en paciente coinfectados con VIH/VHB que no están recibiendo de manera concomitante un tratamiento efectivo para el VIH (ver Precauciones generales / advertencias y precauciones específicas al producto / Coinfección con VIH). Experiencia posterior a la comercialización: Se han identificado los siguientes eventos durante el uso de BARACLUDE® posterior a su aprobación. Como los reportes son realizados voluntariamente por una población de tamaño desconocido, no es posible hacer un estimado de la frecuencia. Trastornos del Sistema Inmune: Reacción anafiláctica. Trastornos del metabolismo y nutrición: Se ha reportado acidosis láctica frecuentemente asociada con descompensación hepática, otras condiciones médicas graves o exposición a medicamentos. Los pacientes con cirrosis descompensada pueden estar en un mayor riesgo de desarrollar acidosis láctica. Trastornos hepatobiliares: Aumento de transaminasas. Trastornos de la piel y del tejido subcutáneo: alopecia, exantema.
Interacciones medicamentosas y de otro género: Productos medicinales: Debido a que el entecavir se elimina predominantemente por vía renal (ver Farmacocinética y farmacodinamia en humanos: metabolismo y eliminación), la coadministración de BARACLUDE® con productos medicinales que reducen la función renal o compiten por la secreción tubular activa puede incrementar las concentraciones séricas de cualquiera de los productos medicinales. La coadministración de BARACLUDE® con lamivudina, dipivoxilo de adefovir o fumarato de disoproxilo de tenofovir no tuvo ninguna interacción farmacológica. No se han evaluado los efectos de la coadministración de BARACLUDE® con otros productos medicinales que se excretan por vía renal o que afectan la función renal. Cuando se coadministra BARACLUDE® con tales productos medicinales, se debe mantener a los pacientes bajo monitoreo estricto para detectar eventos adversos. Alimentos: La administración de entecavir con alimentos disminuyó su absorción en 18-20% (Ver Dosis y vía de admnistración y Absorción).
Alteraciones en los resultados de pruebas de laboratorio: La TABLA 6 muestra los resultados de laboratorio de cuatro estudios clínicos doble ciego, controlados con lamivudina en donde 679 pacientes sin tratamiento previo con nucleósidos recibieron 0.5 mg de BARACLUDE® una vez al día durante una mediana de 54 semanas y 183 pacientes resistentes a lamivudina que recibieron 1 mg de BARACLUDE® durante una mediana de 69 semanas.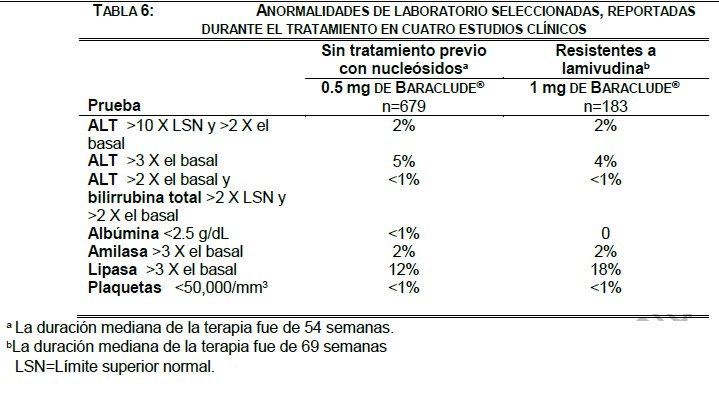
Entre los pacientes tratados con Baraclude®, de estos estudios las elevaciones de ALT > 10 X LSN y > 2 X por encima de la basal que ocurrieron durante el tratamiento, se resolvieron en general con el tratamiento continuo. La mayoría de estas exacerbaciones se asociaron con una reducción de ≥2 log10 /mL en la carga viral que precedió o coincidió con la elevación en ALT. Se recomienda mantener la función hepática bajo monitoreo periódico durante el tratamiento. Anormalidades en las pruebas de laboratorio: Durante las 48 semanas, ninguno de los pacientes tratados con BARACLUDE® presentó elevaciones de ALT > 10 veces al LSN ó > 2 veces los niveles basales, y el 1% de los pacientes presentó elevaciones de ALT > 2 veces los niveles basales junto con bilirrubina total > 2 veces el LSN y > 2 veces los niveles basales. En el 30% de los pacientes los niveles de albúmina fueron < 2.5 g/dL, el 10% de los pacientes tuvieron niveles de lipasa > 3 veces el nivel basal y en el 20% de los pacientes los niveles plaquetarios fueron < 50,000/mm3.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Carcinogénesis, mutagenésis y alteraciones de la fecundidad: Se realizaron estudios de carcinogénesis a dos años con entecavir en ratones y ratas. En los ratones macho, se observó un incremento en la incidencia de tumores en el pulmón con exposiciones a entecavir ≥5 veces la de los humanos que reciben 0.5 mg/día (≥ 3 x la exposición a razón de 1 mg/día). El desarrollo de los tumores fue precedido por proliferación neumocística en el pulmón, lo que no se observó en ratas, perros o monos, respaldando así la conclusión de que los tumores en el pulmón observados en ratones son eventos específicos de la especie y no son relevantes para los humanos. El incremento en la incidencia de otros tipos de tumores relacionados con los medicamentos, incluyendo carcinomas de hígado en ratones macho, tumores vasculares benignos en ratones hembra, gliomas cerebrales en ratas macho y hembra, así como adenomas y carcinomas de hígado en ratas hembra, se observaron sólo con altas exposiciones de entecavir [en ratones, aproximadamente 70 veces la exposición en humanos a 0.5 mg/día (aproximadamente 40 veces a 1 mg/día) y en ratas 62 veces (macho) y 43 veces (hembra) la exposición humana a 0.5 mg/día (35 y 24 times, respectivamente, a 1 mg/día)], en consecuencia, es poco probable que estos resultados sobre los tumores sean relevantes para los humanos. No se observó evidencia de genotoxicidad en la prueba de mutagenicidad microbiana de Ames, una prueba de mutaciones genéticas en células de mamíferos y una prueba de transformación con células embrionarias de hámster sirio. Los resultados de un estudio de micronúcleos orales y un estudio de reparación de ADN oral también resultaron negativos. Entecavir fue clastogénico para cultivos de linfocitos humanos a razón de ≥2350 veces la Cmáx en humanos a 0.5 mg/día (aproximadamente 1200 veces a 1 mg/día). En estudios toxicológicos de entecavir en roedores y perros, se observó degeneración en los túbulos seminíferos a ≥62 y ≥35 veces la exposición en humanos a 0.5 y 1 mg/día, respectivamente. Durante un estudio de 1 año en monos a exposiciones 296 veces la exposición humana a 0.5 mg/día (167 veces a 1 mg/día) no hubo cambios testiculares evidentes. No hubo efectos sobre la fecundidad en ratas macho a exposiciones > 160 veces la exposición humana a 0.5 mg/día ( > 90 veces a 1 mg/día). En ratas hembra, no se observaron efectos sobre la fecundidad ni desarrollo embrionario temprano a exposiciones > 165 veces la exposición humana a 0.5 mg/día ( > 94 veces a 1 mg/día). Efectos teratogénicos: En un estudio de toxicidad del desarrollo en
