BLAXITEC
FAES FARMA
Solución
Denominación genérica: Bilastina
Forma farmacéutica y formulación: Cada 100 mL de solución contiene: Bilastina 250 mg. Excipientes cbp 100 mL. Contiene 0.05% de otros azúcares.
Indicaciones terapeúticas: Tratamiento sintomático de rinitis alérgica (internamente y persistente) y urticaria.
Farmacocinética y farmacodinamia: propiedades farmacodinámicas: Grupo farmacoterapéutico: Antihistamínicos de uso sistemático, otros antihistamínicos de uso sistémico. Código ATC: RO6AX29. Mecanismo de acción: Bilastina es un antagonista no sedante de la histamina de acción prolongada, con afinidad antagonista selectiva por los receptores H1 periféricos y sin afinidad por los receptores muscarínicos. Tras la administración de dosis única; bilastina inhibió durante 24 horas las manifestaciones cutáneas de urticaria y eritema inducidas por histamina. Propiedades farmacocinéticas: Bilastina presenta una farmacocinética lineal en el rango de dosis estudiada (5 a 220 mg), con una muy baja variabilidad inter individual. Bilastina se absorbe rápidamente tras la administración oral hasta alcanzar la concentración plasmática máxima de aproximadamente 1.3 horas. No se ha observado acumulación. El valor medio de la biodisponibilidad es de 61%. Estudios in vitro e in vivo han demostrado que bilastina es un substrato de la P-gp y de la OATP. A las dosis terapéuticas la unión de bilastina a las proteínas plasmáticas es de 84-90%. En estudios in vitro bilastina no indujo ni inhibió la actividad de las isoenzimas del CYP450. En un estudio de balance de masas realizado en voluntarios sanos, tras la administración de una dosis única de 20 mg de 14C-Bilastina, casi el 95% de la dosis administrada fue recuperada en orina (28.3%) y heces (66.5%) como bilastina inalterada, confirmando que bilastina no es significativamente metabolizada en humanos. La vida media de eliminación calculada en voluntarios sanos fue de 14.5 h. Pacientes con Insuficiencia renal: Los efectos de bilastina en pacientes con insuficiencia renal se han estudiado en adultos. En un estudio realizado en sujetos con insuficiencia renal la AUC0-inf media (DE) aumentó de 737.4 (±260.8) ngxhr/mL en sujetos sin insuficiencia (IFG: > 80 mL/min/1.73 m²) a: 967.4 (±140.2) ngxhr/mL en sujetos con insuficiencia leve (IFG: 50-80 mL/min/1.73 m²), 1 384.2 (±263.23) ngxhr/mL en sujetos con insuficiencia moderada (lFG: 30- < 50 mL/min/1.73 m²) y 1 708.5 (±699.0) ngxhr/mL en sujetos con insuficiencia severa (IFG: < 30 mL/min/1.73 m²). La vida media de eliminación (media±DE) de bilastina fue de 9.3 h (±2.8) en sujetos con función renal normal, 15.1 h (±7.7) en sujetos con insuficiencia leve, 10.5 h (±2.3) en sujetos con insuficiencia moderada y 18.4 h (±11.4) en sujetos con insuficiencia severa. La excreción urinaria de bilastina fue completa tras 48-72 h en todos los sujetos. No cabe esperar que estos cambios farmacocinéticos tengan una influencia clínicamente relevante sobre la seguridad de bilastina, ya que los niveles plasmáticos de bilastina en pacientes con insuficiencia renal continúan estando dentro del rango de seguridad de bilastina. Pacientes con Insuficiencia hepática: No hay datos farmacocinéticos en sujetos con insuficiencia hepática. Bilastina no es metabolizada en humanos. Puesto que los resultados del estudio en insuficiencia renal indican que la vía renal es la principal responsable de la eliminación cabe esperar que la excreción biliar solo este implicada de forma marginal en la eliminación de bilastina. No se espera que los cambios en la función hepática tengan una influencia clínicamente relevante en la farmacocinética de bilastina. Pacientes pediátricos: Los datos farmacocinéticos en niños se obtuvieron en un estudio farmacocinético de Fase II, que incluyó 31 niños de 4-11 años de edad con diagnóstico clínico de rinoconjuntivitis alérgica o urticaria crónica, a los que se administró una solución de bilastina 10 mg una vez al día. Esta formulación ha demostrado ser bioequivalente a bilastina 2.5 mg/mL solución. El modelo farmacocinético de los datos de concentración plasmática mostró que la dosis pediátrica de 10 mg de bilastina una vez al día da lugar a una exposición sistémica equivalente a la observada después de una dosis de 20 mg en adultos y adolescentes. Los valores en estado estacionario de Cmax y AUC fueron aproximadamente de 229 ng/mL y 783 ng*h/mL, respectivamente. Estos resultados estuvieron ampliamente por debajo del umbral de seguridad en base a los datos de dosis de 80 mg una vez al día en adultos, de acuerdo con el perfil de seguridad del fármaco. La influencia de la edad sobre los principales parámetros farmacocinéticos de bilastina (CL/F y V/F) no fue significativa en la población de estudio, por lo tanto, la dosis empleada y los resultados del estudio también son aplicables a edades más jóvenes. Estos resultados confirmaron la elección de la bilastina 10 mg vía oral una vez al día como la dosis terapéutica apropiada para la población pediátrica en el rango de edad de 2-11 años. Eficacia clínica: La eficacia de bilastina ha sido estudiada y demostrada en adultos y adolescentes. La eficacia demostrada en adultos y adolescentes se puede extrapolar a niños, habiéndose probado que la exposición sistémica con 10 mg de bilastina en niños de 2-11 años es equivalente a la exposición en adultos con 20 mg de bilastina. La extrapolación de los datos de adultos y adolescentes se considera apropiada para este medicamento ya que la fisiopatología de la rinitis alérgica y urticaria es la misma para todos los grupos de edad. En los ensayos clínicos realizados en pacientes adultos y adolescentes con rinitis alérgica (intermitente y persistente), bilastina administrada en dosis única diaria de 20 mg durante 14-28 días, fue eficaz para aliviar los síntomas, tales como estornudos, rinorrea, picor nasal, congestión nasal, prurito ocular, lagrimeo y enrojecimiento ocular. Bilastina controló los síntomas de forma eficaz durante 24 horas. En dos ensayos clínicos realizados en pacientes con urticaria crónica idiopática, bilastina administrada en dosis única diaria de 20 mg durante 28 días fue eficaz para aliviar la intensidad del prurito y el número y tamaño de las ronchas, así como el malestar de los pacientes derivado de la urticaria. Los pacientes obtuvieron una mejoría en la calidad del sueño y en la calidad de vida. En los ensayos clínicos realizados con bilastina no se observó ninguna prolongación del intervalo QTc ni ningún otro efecto cardiovascular clínicamente relevantes, incluso a dosis de hasta 200 mg diarios (10 veces la dosis terapéutica) durante 7 días en 9 sujetos, o incluso cuando se administraron de forma concomitante inhibidores de P-gp, tales como ketoconazol (24 sujetos) y eritromicina (24 sujetos). Además, se ha llevado a cabo un estudio de profundidad de análisis del QT en 30 voluntarios. En los ensayos clínicos controlados realizados con la dosis recomendada, 20 mg una vez al día, el perfil de seguridad de bilastina sobre SNC fue similar al del placebo y la incidencia de somnolencia no fue estadísticamente diferente a placebo. Bilastina a dosis hasta 40 mg q.d. no afectó el rendimiento psicomotor en los ensayos clínicos y no afectó a la capacidad de conducción en un estudio estándar sobre desempeño en una prueba de manejo. La eficacia y seguridad de bilastina en los pacientes de edad avanzada (≥65 años) incluidos en los estudios de fase II y III no mostraron diferencias significativas con respecto a los pacientes más jóvenes. Seguridad clínica: En un ensayo clínico controlado de 12 semanas con niños de 2 a 11 años de edad (509 niños en total, 260 tratados con 10 mg de bilastina: 58 de 2 a < 6 años de edad, 105 de 6 a < 9 años de edad y 97 de 9 a < 12 años de edad; y 249 tratados con placebo: 58 de 2 a < 6 años de edad, 95 de 6 a < 9 años de edad y 96 de 9 a < 12 años de edad), a la dosis pediátrica recomendada de 10 mg una vez al día, el perfil de seguridad de bilastina (n =260) fue similar a placebo (n =249), con reacciones adversas a medicamentos observados en 5.8% y 8.0% de los pacientes que tomaron 10 mg de bilastina y placebo, respectivamente. Durante este estudio, ambos grupos, bilastina 10 mg y placebo, mostraron una ligera disminución en las puntuaciones de somnolencia y sedación en el Cuestionario de Sueño Pediátrico, sin diferencias estadísticamente significativas entre los grupos de tratamiento. En estos niños de 2-11 años de edad no se detectaron diferencias significativas en QTc tras la administración de 10 mg de bilastina diaria en comparación con placebo. Los cuestionarios de Calidad de Vida específicos para niños con rinoconjuntivitis alérgica o urticaria crónica mostraron un incremento general en las puntuaciones tras 12 semanas sin diferencia estadísticamente significativa entre los grupos de bilastina y placebo. La población total de 509 niños incluyó: 479 sujetos con rinoconjuntivitis alérgica y 30 sujetos diagnosticados de urticaria crónica. 260 niños recibieron bilastina, 252 (96.9%) para rinoconjuntivitis alérgica y 8 (3.1%) para la urticaria crónica. Por analogía, 249 niños recibieron placebo, 227 (91.2%) para rinoconjuntivitis alérgica y 22 (8.8%) para la urticaria crónica.
Contraindicaciones: Hipersensibilidad a la sustancia activa de bilastina o a alguno de los excipientes.
Precauciones generales: Debe tomarse en condiciones de ayuno una hora antes o dos horas después de la ingesta de alimento o jugo de frutas (ver sección Interacciones medicamentosas y de otro género). La eficacia y seguridad de bilastina en niños menores de 2 años no se ha establecido. Se recomienda precaución cuando bilastina se co administre con inhibidores de P-glicoproteína tales como ketoconazol, eritromicina, ciclosporina, ritonavir o diltiazem, ya que en pacientes con insuficiencia renal moderada o severa, la administración concomitante de bilastina con estos inhibidores puede aumentar los niveles plasmáticos de bilastina y por lo tanto aumentar el riesgo de reacciones adversas de bilastina. Por ello, la administración concomitante de bilastina e inhibidores de la glicoproteína P debe evitarse en pacientes con insuficiencia renal moderada o severa. En pacientes con insuficiencia renal se deberá tener cuidado con el uso de bilastina, sobre todo en edades pediátricas. En pacientes con insuficiencia renal severa no se recomienda el uso de bilastina debido a que no se tiene la evidencia en este tipo de pacientes. Efectos sobre la capacidad para manejar vehículos de motor o utilizar maquinaria. Un estudio realizado para evaluar los efectos de bilastina sobre la capacidad de conducción demostró que el tratamiento con 20 mg no afectó el desempeño durante la prueba de manejo. No obstante, se debe informar a los pacientes de que muy raramente algunas personas experimentan somnolencia, lo que puede afectar a su capacidad para conducir o utilizar máquinas. Como medida de precaución, es preferible evitar el uso de Blaxitec® durante el embarazo. Lactancia: Se debe decidir si continuar/discontinuar la lactancia o interrumpir/abstenerse del tratamiento con Blaxitec® tras considerar el beneficio de la lactancia para el niño y el beneficio del tratamiento con bilastina para la madre.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: No hay datos o estos son limitados relativos al uso de bilastina en mujeres embarazadas. Los estudios en animales no sugieren efectos perjudiciales directos ni indirectos en términos de toxicidad para la reproducción, el parto o el desarrollo postnatal (ver sección Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad); como medida de precaución, es preferible evitar el uso de bilastina durante el embarazo. Lactancia: La excreción de bilastina en la leche no ha sido estudiada en humanos. Los datos farmacocinéticos disponibles en animales muestran que bilastina se excreta en la leche. Se debe decidir si es preferible continuar/interrumpir la lactancia o continuar/interrumpir el tratamiento con bilastina tras considerar el beneficio de la lactancia para el niño y el beneficio del tratamiento para la madre.
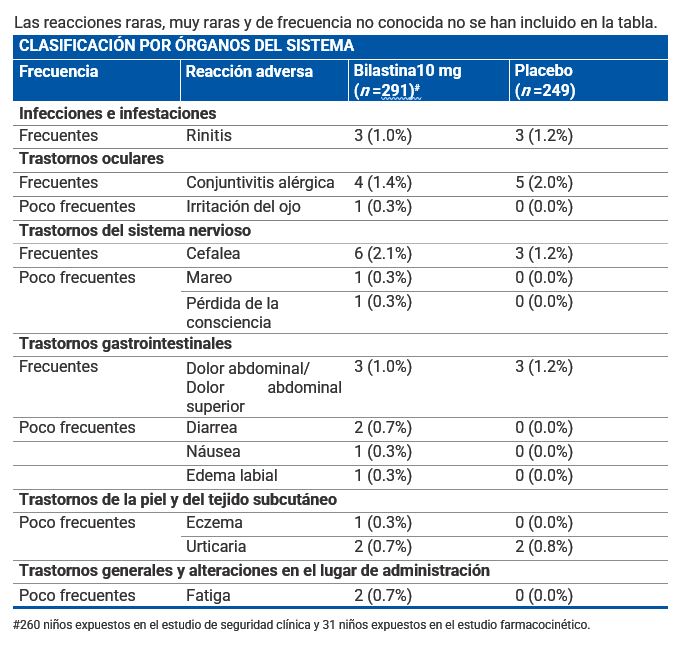
Reacciones secundarias y adversas: Población pediátrica: Durante el desarrollo clínico la frecuencia, tipo y severidad de las reacciones adversas en adolescentes (12-17 años) fueron los mismos que en adultos. La información obtenida en esta población durante la vigilancia post-comercialización ha confirmado lo acontecido durante los ensayos clínicos. El porcentaje de niños (2-11 años) que informaron de eventos adversos (EAs) después del tratamiento con bilastina 10 mg para la rinitis alérgica o urticaria crónica idiopática en un ensayo clínico controlado de 12 semanas, fue comparable con los pacientes que recibieron placebo (68.5% frente a 67.5%). Los acontecimientos adversos más comúnmente reportados por 291 niños (2-11 años) que reciben bilastina durante los ensayos clínicos (formulación de solución) fueron cefalea, dolor abdominal, conjuntivitis alérgica y rinitis. Estos eventos adversos se produjeron con una frecuencia comparable en 249 pacientes que recibieron placebo. La siguiente tabla muestra los EAs al menos posiblemente relacionados con bilastina y notificados en más del 0.1% de los niños (2-11 años) tratados con bilastina durante el desarrollo clínico. Las frecuencias se han clasificado de la siguiente forma: Muy frecuentes (≥1/10) Frecuentes (≥1/100 a < 1/10). Poco frecuentes (≥1/1 000 a < 1/100) Raras (≥1/10 000 a < 1/1 000). Muy raras ( < 1/10 000). Frecuencia no conocida (no puede estimarse a partir de los datos disponibles): Las reacciones raras, muy raras y de frecuencia no conocida no se han incluido en la tabla.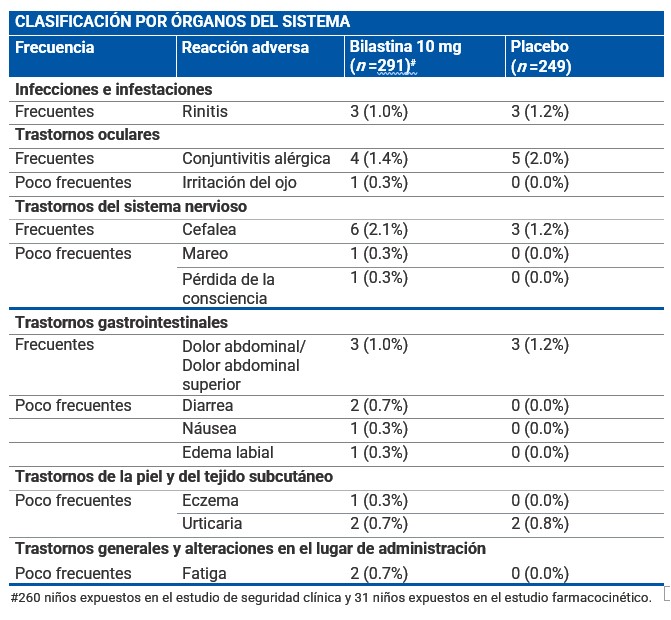
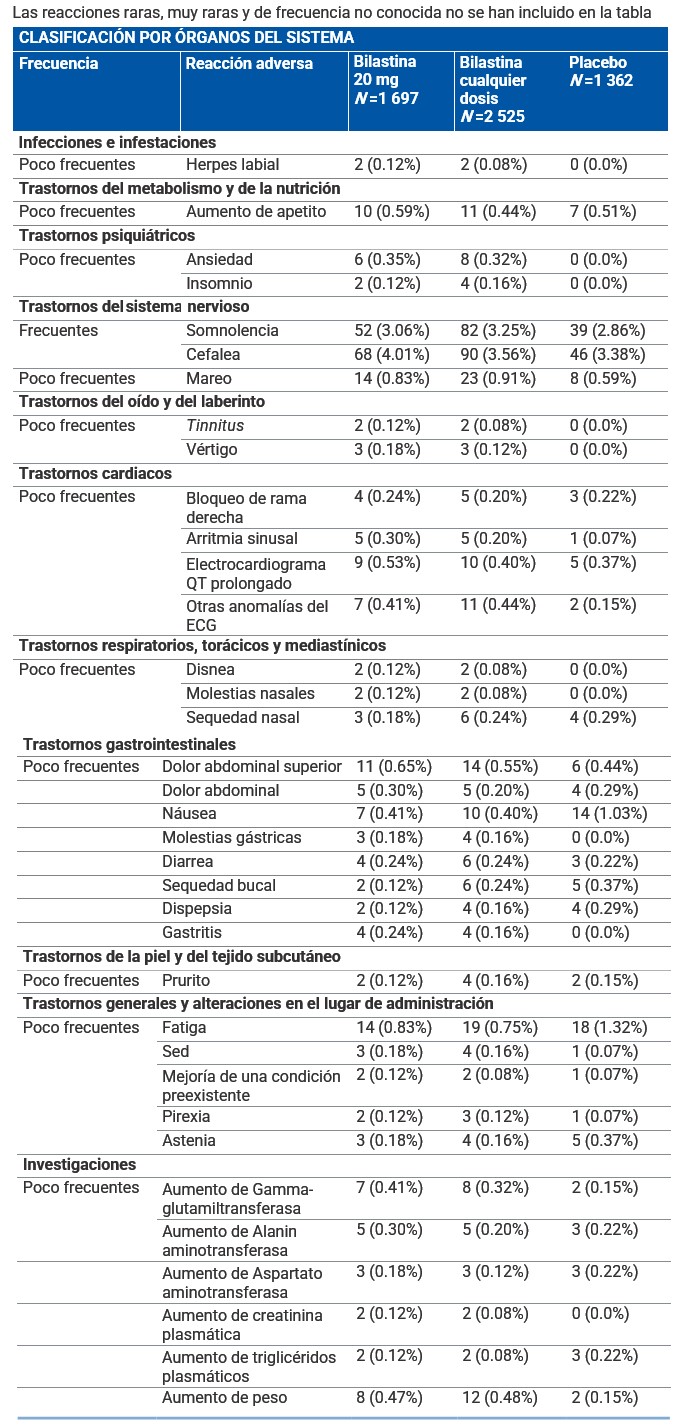
Descripción de las reacciones adversas encontradas en la población pediátrica: El dolor de cabeza, dolor abdominal, la conjuntivitis alérgica y la rinitis se observaron tanto en niños tratados con bilastina 10 mg como con placebo. La frecuencia reportada fue de 2.1% frente a 1.2% para el dolor de cabeza; 1.0% vs. 1.2% para el dolor abdominal; 1.4% frente a 2.0% para la conjuntivitis alérgica y 1.0% frente a 1.2% para la rinitis. Adolescentes y adultos: La incidencia de eventos adversos en los pacientes afectados de rinitis alérgica o urticaria crónica idiopática tratados con bilastina 20 mg en los estudios clínicos fue comparable a la incidencia en pacientes que recibieron placebo (12.7% frente a 12.8%). Los ensayos clínicos de fase II y III realizados durante el desarrollo clínico incluyeron 2 525 pacientes adultos y adolescentes tratados con diferentes dosis de volatina, de los cuales, 1 697 recibieron 20 mg de bilastina. Adicionalmente, en estos ensayos 1 362 pacientes recibieron placebo. Las reacciones adversas notificadas más frecuentemente por los pacientes tratados con bilastina 20 mg para la indicación de rinitis alérgica o urticaria crónica idiopática fueron cefalea, somnolencia, mareo y fatiga. Estos eventos adversos ocurrieron con una frecuencia similar en los pacientes que recibieron placebo. La siguiente tabla muestra las reacciones adversas al menos posiblemente relacionadas con bilastina y notificadas en más del 0.1% de los pacientes tratados con bilastina 20 mg durante el desarrollo clínico (N =1 697). Las frecuencias se han clasificado de la siguiente forma: Muy frecuentes (≥1/10) Frecuentes (≥1/100 a < 1/10). Poco frecuentes (≥1/1 000 a < 1/100) Raras (≥1/10 000 a < 1/1 000). Muy raras ( < 1/10 000). Frecuencia no conocida (no puede estimarse a partir de los datos disponibles): Las reacciones raras, muy raras y de frecuencia no conocida no se han incluido en la tabla.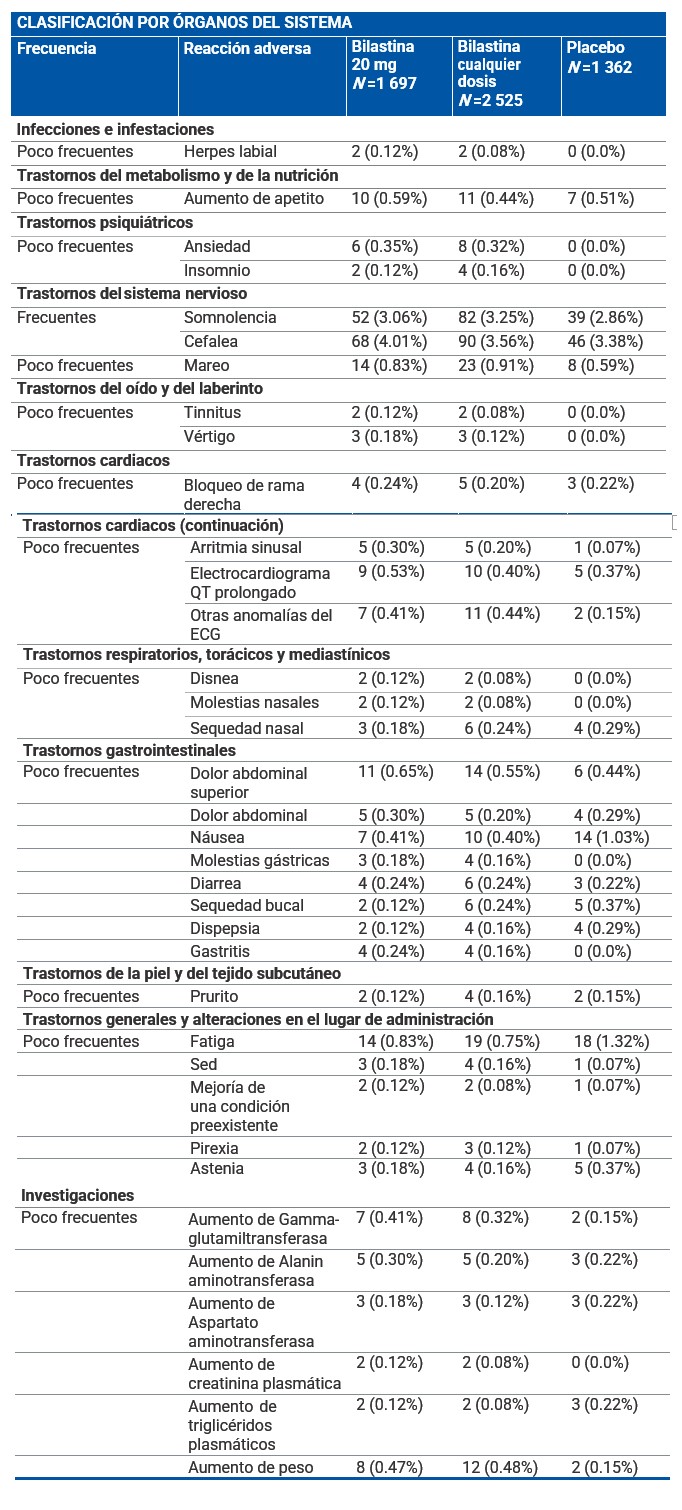
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles): Se han observado palpitaciones, taquicardia y reacciones de hipersensibilidad (como anafilaxia, angioedema, disnea, erupción cutánea, edema localizado/hinchazón local y eritema) durante el periodo de postcomercialización. Descripción de las reacciones adversas relevantes: Se observó somnolencia, cefalea, mareo y fatiga tanto en pacientes tratados con bilastina 20 mg como con placebo. La frecuencia notificada fue de 3.06% vs. 2.86% para somnolencia; 4.01% vs. 3.38% para cefalea; 0.83% vs. 0.59% para mareo y 0.83% vs. 1.32% para fatiga. La información recogida durante la vigilancia postcomercialización ha confirmado el perfil de seguridad observado durante el desarrollo clínico.
Interacciones medicamentosas y de otro género: Los estudios de interacciones se han realizado solo en adultos y se resumen a continuación: Interacción con alimentos: Los alimentos reducen significativamente la biodisponibilidad oral de bilastina en un 30% y de bilastina 2.5 mg/mL solución en un 20%. Interacción con jugo de toronja: La administración concomitante de bilastina 20 mg y jugo de toronja disminuyó la biodisponibilidad de bilastina en un 30%. Este efecto puede ocurrir también con otros jugos o derivados de las frutas. El grado de reducción en la biodisponibilidad puede variar entre fabricantes y frutos. El mecanismo responsable de esta interacción es la inhibición del OATP1A2, un transportador de captación del cual bilastina es sustrato. Los medicamentos que sean sustratos o inhibidores del OATP1A2, tales como ritonavir o rifampicina, podrían igualmente reducir las concentraciones plasmáticas de bilastina. Interacción con ketoconazol o eritromicina: La administración concomitante de bilastina 20 mg una vez al día y ketoconazol 400 mg una vez al día o eritromicina 500 mg 3 veces al día aumentó el AUC de bilastina en 2 veces y la Cmax en 2-3 veces. Estos cambios se pueden explicar debido a la interacción con trasportadores intestinales de excreción, ya que bilastina es sustrato de la P-gp y no es metabolizada. Estos cambios no parecen afectar al perfil de seguridad de bilastina y ketoconazol o eritromicina, respectivamente. Otros medicamentos que sean inhibidores de la P-gp, tal como ciclosporina, podrían igualmente aumentar las concentraciones plasmáticas de bilastina. Interacción con diltiazem: La administración concomitante de bilastina 20 mg y diltiazem 60 mg aumenta la Cmax de bilastina en un 50%. Este efecto se puede explicar por la interacción con transportadores intestinales de excreción y no parece afectar al perfil de seguridad de bilastina. Interacción con alcohol: El rendimiento psicomotor después de la toma concomitante de alcohol y 20 mg de bilastina fue similar tras la administración del alcohol y placebo. Interacción con lorazepam: La administración concomitante de bilastina 20 mg y lorazepam 3 mg por 8 días no potenció los efectos depresores del SNC causados por lorazepam.
Alteraciones en los resultados de pruebas de laboratorio: No se observó ninguna desviación clínica ni estadísticamente significativa asociada al tratamiento con bilastina en ninguno de los laboratorios estudiados.
Precauciones en relación con efecto de carcinogénesis, mutagénesis, teratogénesis y sobre fertilidad: Los datos de estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas, genotoxicidad y potencial carcinogénico. En los estudios de toxicidad para la reproducción únicamente se observaron efectos de bilastina sobre el feto (pérdidas pre y post implantación en plantas y osificación incompleta de huesos craneales, esternón y miembros en conejos) a dosis tóxicas para la madre. Los niveles de exposición determinados por las NOAEL son superiores ( > 30 veces) a los niveles de exposición alcanzados en humanos a la dosis terapéutica recomendada. En un estudio sobre lactancia se detectó bilastina en la leche de ratas en periodo de lactancia tras la administración oral de una dosis única (20 mg/kg). Las concentraciones de bilastina en la leche fueron alrededor de la mitad de las del plasma materno. Se desconoce la relevancia de estos resultados para los humanos. En un estudio de fertilidad en ratas, la administración oral de bilastina a dosis hasta 100 mg/kg/día no indujo ningún efecto sobre los órganos reproductivos de los machos ni de las hembras. Los índices de apareamiento, fertilidad y gravidez no se vieron afectados. Tal y como se observó en un estudio de distribución en ratas con determinación de las concentraciones de fármaco por técnicas de medicina nuclear, bilastina no se acumula a nivel del SNC.
Dosis y vía de administración: Niños de 2-11 años de edad: 10 mg de bilastina (4 mL de solución) una vez al día. Blaxitec® 2.5 mg/mL solución debe tomarse una hora antes o dos horas después de la ingesta de alimentos o jugos de frutas. Poblaciones especiales: Pacientes con insuficiencia renal: No se ha establecido la seguridad y eficacia de la bilastina en niños con insuficiencia renal. Los estudios realizados en adultos en grupos especiales de riesgo (pacientes con insuficiencia renal) indican que no es necesario ajustar la dosis de bilastina en adultos. Pacientes con insuficiencia hepática: No hay experiencia clínica en pacientes con insuficiencia hepática. Como bilastina no se metaboliza y el aclaramiento renal es su principal vía de eliminación, no se espera que la insuficiencia hepática aumente la exposición sistémica por encima del margen de seguridad. Por lo tanto, no se requiere ajuste de dosis en pacientes con insuficiencia hepática. Duración del tratamiento: Para rinitis alérgica el tratamiento debe limitarse al periodo de exposición a los alérgenos. Para rinitis alérgica intermitente el tratamiento puede interrumpirse cuando se hayan resuelto los síntomas y reiniciarse en caso de que estos reaparezcan. En rinitis alérgica persistente se puede proponer al paciente el tratamiento continuado durante los periodos de exposición a los alérgenos. Para urticaria la duración del tratamiento depende del tipo, duración y evolución de los síntomas. En los ensayos clínicos controlados realizados con la dosis recomendada de 20 mg una vez al día, el perfil de seguridad de bilastina sobre el SNC fue similar al placebo y la incidencia de somnolencia no fue estadísticamente diferente a placebo. Bilastina a dosis hasta 40 mg al día no afectó al rendimiento psicomotor en los ensayos clínicos y no afectó a la capacidad de conducción en un estudio estándar de conducción. Método de administración: Vía oral. La solución se acompaña de un vasito para dosificar con una marca de 4 mL =10 mg de bilastina por dosis.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: No existen datos acerca de la sobredosis en niños. La información relacionada con sobredosis aguda de bilastina se recoge de la experiencia de los estudios clínicos realizados durante el desarrollo en adultos y durante la postcomercialización. En los ensayos clínicos, tras la administración de bilastina a dosis de 10 a 11 veces la dosis terapéutica (220 mg como dosis única o 200 mg/día durante 7 días) a 26 voluntarios sanos, la frecuencia de eventos adversos tras el tratamiento fue dos veces superior a la observada tras la administración de placebo. Las reacciones adversas más frecuentemente notificadas fueron mareo, cefalea y náusea. No se notificaron eventos adversos serios ni prolongaciones significativas del intervalo QTc. La información recogida durante la post-comercialización coincide con la información obtenida en los ensayos clínicos. La evaluación crítica del efecto de dosis múltiple de bilastina (100 mg durante 4 días) sobre la repolarización ventricular en un estudio cruzado de "Análisis profundo del QT/QTc" realizado con 30 voluntarios sanos no mostró ninguna prolongación significativa del intervalo QTc. En caso de producirse una sobredosis se recomienda tratamiento sintomático y de soporte. No se conoce ningún antídoto específico para bilastina
Presentaciones: Caja con frasco de vidrio ámbar, sellado con cápsula de aluminio, incluyendo un vasito dosificador graduado de 4 mL. Cada frasco contiene 120 mL.
Recomendaciones sobre el almacenamiento: Consérvese a temperatura ambiente a no más de 30 °C y en un lugar seco.
Leyendas de protección: Su venta requiere receta médica. No se deje al alcance de los niños. Literatura exclusiva para médicos. No se administre en el embarazo y lactancia. No se administre a menores de 2 años de edad. Consérvese el frasco bien cerrado. Una vez abierto, el producto se conserva máximo durante 6 meses. Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx o al correo: farmacovigilancia@faesfarma.com.mx En caso de dudas comunicarse al 55 56 61 1071.
Nombre y domicilio del laboratorio: Hecho en España por: FAES FARMA, S.A. C/Máximo Aguirre, 14, Lejona-Leioa, 48940, Vizcaya. España. Representante legal e importado por: FAES FARMA MÉXICO S.A. de C.V. Avenida Prolongación Paseo de la Reforma 51 Piso 8, Oficina 802, Col. Paseo de las Lomas, C.P. 01330, Del. Álvaro Obregón, Ciudad de México, México. Importado y Distribuido por: FAES FARMA MÉXICO S.A. de C.V. Avenida Gustavo Baz No. 109, Puerta 16-A. Col. San Pedro Barrientos, C.P. 54010. Tlalnepantla de Baz, México, México.
No. de registro del medicamento: 263M2018 SSA IV.
BLAXITEC
FAES FARMA
Tabletas
Denominación genérica: Bilastina
Forma farmacéutica y formulación: Cada tableta contiene: Bilastina 20 mg. Excipiente cbp. 1 tableta
Indicaciones terapéuticas: Tratamiento sintomático de la rinoconjuntivitis alérgica (intermitente y perenne) y de la urticaria. Blaxitec 20 mg comprimidos está indicado en adultos y adolescentes (edad igual o superior a 12 años).
Farmacocinética y farmacodinamia: Propiedades farmacocinéticas: Absorción: Bilastina se absorbe rápidamente tras la administración oral con un tiempo hasta alcanzar la concentración plasmática máxima de aproximadamente 1.3 horas. No se ha observado acumulación. La biodisponibilidad oral media de bilastina es del 61%. Distribución: Estudios in vitro e in vivo han demostrado que bilastina es un substrato de la P-gp y del OATP. Bilastina no parece ser un substrato del transportador BCRP ni de los transportadores renales OCT2, OAT1 y OAT3. En base a los estudios in vitro, no cabe esperar que bilastina inhiba los siguientes transportadores a nivel de la circulación sistémica: P-gp, MRP2, BCRP, BSEP, OATP1B1, OATP1B3, OATP2B1, OAT1, OAT3, OCT1, OCT2, y NTCP, ya que solo se detectó una ligera inhibición para P-gp, OATP2B 1 y OCT1, estimándose una CI50 ≥300 mM, muy superior a la concentración plasmática máxima (Cmax) y por ello, estas interacciones carecen de relevancia clínica. Sin embargo, en base a estos resultados no se puede descartar que bilastina sea inhibidor de transportadores presentes en la mucosa intestinal, como por ejemplo P-gp. A las dosis terapéuticas la unión de bilastina a las proteínas plasmáticas es de 84-90%. Metabolismo o Biotransformación: En estudios in vitro bilastina no indujo ni inhibió la actividad de los isoenzimas del CYP450. Eliminación: En un estudio de balance de masas realizado en voluntarios sanos, tras la administración de una dosis única de 20 mg de 14C-bilastina, casi el 95% de la dosis administrada fue recuperada en orina (28.3%) y heces (66.5%) como bilastina inalterada, confirmando que bilastina no es significativamente metabolizada en humanos. La vida media de eliminación calculada en voluntarios sanos fue de 14.5 h. Linealidad/No linealidad: Bilastina presenta una farmacocinética lineal en el rango de dosis estudiado (5 a 220 mg), con una baja variabilidad interindividual. Insuficiencia renal: En un estudio realizado en sujetos con insuficiencia renal la AUC0-∞ media (DE) aumentó de 737.4 (±260.8) ngxhr/mL en sujetos sin insuficiencia (TFG: > 80 mL/min/1.73 m2) a: 967.4 (±140.2) ngxhr/mL en sujetos con insuficiencia leve (IFG: 50-80 mL/min/1.73 m2), 1 384.2 (±263.23) ngxhr/mL en sujetos con insuficiencia moderada (TFG: 30 - < 50 mL/min/1.73 m2), y 1 708.5 (±699.0) ngxhr/mL en sujetos con insuficiencia severa (TFG: < 30 mL/min/1. 73 m2). La vida media de eliminación (media ± DE) de bilastina fue de 9.3 h (±2.8) en sujetos con función renal normal, 15.1 h (±7.7) en sujetos con insuficiencia leve, 10.5 h (±2.3) en sujetos con insuficiencia moderada y 18.4 h (±11.4) en sujetos con insuficiencia severa. La excreción urinaria de bilastina fue completa tras 48-72 h en todos los sujetos. No cabe esperar que estos cambios farmacocinéticos tengan una influencia clínicamente relevante sobre la seguridad de bilastina, ya que los niveles plasmáticos de bilastina en pacientes con insuficiencia renal continúan estando dentro del rango de seguridad de bilastina. Insuficiencia hepática: No hay datos farmacocinéticos en sujetos con insuficiencia hepática. Bilastina no es metabolizada en humanos. Puesto que los resultados del estudio en insuficiencia renal indican que la vía renal es la principal responsable de la eliminación cabe esperar que la excreción biliar solo esté implicada de forma marginal en la eliminación de bilastina. No se espera que los cambios en la función hepática tengan una influencia clínicamente relevante en la farmacocinética de bilastina. Pacientes de edad avanzada: En sujetos mayores de 65 años solo se dispone de datos farmacocinéticos limitados. No se han observado diferencias estadísticamente significativas en la farmacocinética de bilastina en sujetos de edad avanzada (mayores de 65 años) comparados con la población adulta de edad comprendida entre 18 y 35 años. Propiedades farmacodinámicas: Grupo farmacoterapéutico: Antihistamínicos de uso sistémico, otros antihistamínicos de uso sistémico. Código ATC: RO6AX29. Bilastina es un antagonista no sedante de la histamina de acción prolongada, con afinidad antagonista selectiva por los receptores H1 periféricos y sin afinidad por lo receptores muscarínicos. Tras la administración de dosis única; bilastina inhibió durante 24 horas las manifestaciones cutáneas de urticaria y eritema inducidas por histamina. En estudios clínicos realizados en pacientes adultos y adolescentes con rinoconjuntivitis alérgica (estacional y persistente), bilastina administrada en una única dosis diaria de 20 mg durante 14-28 días fue eficaz para aliviar los síntomas, tales como estornudos, rinorrea, picor nasal, congestión nasal, picor ocular, lagrimeo y enrojecimiento ocular. Bilastina controló los síntomas de forma eficaz durante 24 horas. En dos estudios clínicos realizados en pacientes con urticaria crónica idiopática, bilastina administrada en una única dosis diaria de 20 mg durante 28 días fue eficaz para aliviar la intensidad del prurito y el número y tamaño de las lesiones (ronchas), así como el malestar de los pacientes derivado de la urticaria. Los pacientes obtuvieron una mejoría en la calidad del sueño y en la calidad de vida. En los estudios clínicos realizados con bilastina no se observó ninguna prolongación del intervalo QTc ni ningún otro efecto cardiovascular clínicamente relevantes, incluso a dosis hasta 200 mg diarios (10 veces la dosis terapéutica) durante 7 días en 9 sujetos o incluso cuando se administraron de forma concomitante inhibidores de P-gp, tales como ketoconazol (24 sujetos) y eritromicina (24 sujetos). Además, se ha llevado a cabo un estudio de profundidad de análisis del QT en 30 voluntarios. En los estudios clínicos controlados realizados con la dosis recomendada, 20 mg una vez al día, el perfil de seguridad de bilastina sobre el SNC fue similar al del placebo y la incidencia de somnolencia no fue estadísticamente diferente a placebo. Bilastina a dosis hasta 40 mg q.d. no afectó al rendimiento psicomotor en los estudios clínicos y no afectó a la capacidad de conducción en un estudio estándar sobre desempeño en una prueba de manejo. La eficacia y seguridad de bilastina en los pacientes de edad avanzada (≥65 años) incluidos en los estudios de fase II y III no mostraron diferencias con respecto a pacientes más jóvenes. Un estudio postautorización con 146 pacientes de edad avanzada no mostró diferencias en el perfil de seguridad con respecto a la población adulta.
Contraindicaciones: Hipersensibilidad a la sustancia activa o a alguno de los excipientes.
Precauciones generales: La tableta debe tomarse en condiciones de ayuno una hora antes o dos horas después de la ingesta de alimento o jugo de frutas (Ver sección Interacciones medicamentosas y de otro género). La eficacia y seguridad de bilastina en niños menores de 12 años no se ha establecido. Se recomienda precaución cuando bilastina se coadministra con inhibidores de P-glicoproteína en pacientes con disfunción renal moderada o severa. Se recomienda precaución con el uso de bilastina durante el embarazo o lactancia ya que no existen estudios adecuados y bien controlados de bilastina en estas poblaciones (Ver sección Restricciones de uso durante el embarazo y lactancia). Un estudio realizado para establecer el efecto de bilastina sobre la capacidad de conducción demostró que el tratamiento con 20 mg de bilastina no afecta al rendimiento durante la conducción. Sin embargo, muy raramente algunas personas pueden notar somnolencia, lo que puede afectar a su capacidad para conducir o usar máquinas.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: No hay datos o estos son limitados relativos al uso de bilastina en mujeres embarazadas. Los estudios en animales no sugieren efectos perjudiciales directos ni indirectos en términos de toxicidad para la reproducción, el parto o el desarrollo postnatal (Ver sección Precauciones en relación con efecto de carcinogénesis, mutagénesis y teratogénesis y sobre la fertilidad) como medida de precaución, es preferible evitar el uso de bilastina durante el embarazo. Lactancia: No se ha estudiado en humanos la excreción de bilastina en la leche. Los datos farmacocinéticos disponibles en animales muestran que bilastina se excreta en la leche (Ver sección Precauciones en relación con efecto de carcinogénesis, mutagénesis, teratogénesis y sobre fertilidad). Se debe decidir si es necesario interrumpir o abstenerse del tratamiento con bilastina 20 mg tras considerar el beneficio de la lactancia para el niño y el beneficio del tratamiento para la madre. Fertilidad: No hay datos clínicos o estos son limitados. En un estudio en ratas no se detectó ningún efecto negativo sobre la fertilidad (Ver sección Precauciones en relación con efecto de carcinogénesis, mutagénesis y teratogénesis y sobre la fertilidad).
Reacciones secundarias y adversas: Resumen del perfil de seguridad: La incidencia de eventos adversos en pacientes afectados de rinoconjuntivitis alérgica o urticaria crónica idiopática tratados con bilastina 20 mg en los estudios clínicos fue comparable al observado en los pacientes que recibieron placebo (12.7% frente a 12.8%). Los estudios clínicos de fase II y III realizados durante el desarrollo clínico incluyeron 2 525 pacientes tratados con diferentes dosis de bilastina, de los cuales 1 697 recibieron 20 mg de bilastina. Adicionalmente, en estos ensayos 1 362 pacientes recibieron placebo. Las reacciones adversas notificadas más frecuentemente por los pacientes tratados con bilastina 20 mg para la indicación de rinoconjuntivitis alérgica o urticaria crónica idiopática fueron cefalea, somnolencia, mareo y fatiga. Estos eventos adversos ocurrieron con una frecuencia similar en los pacientes que recibieron placebo. Resumen tabulado de reacciones adversas: La siguiente tabla muestra las reacciones adversas al menos posiblemente relacionadas con bilastina y notificadas en más del 0.1% de los pacientes tratados con bilastina 20 mg durante el desarrollo clínico (N =1 697). Las frecuencias se han clasificado de la siguiente forma: Muy frecuentes (≥1/10) Frecuentes (≥1/100 a < 1/10). Poco frecuentes (≥1/1 000 a < 1/100) Raras (≥1/10.000 a < 1/1.000). Muy raras ( < 1/10.000). Frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones raras, muy raras y de frecuencia no conocida no se han incluido en la tabla.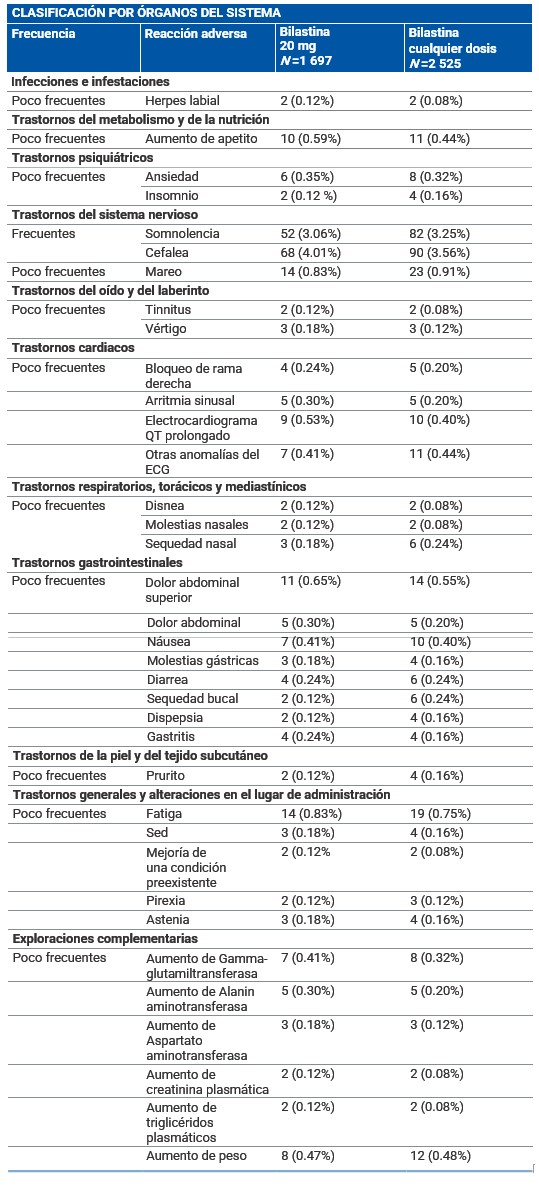
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles): se han observado palpitaciones, taquicardia y reacciones de hipersensibilidad (como anafilaxia, angioedema, disnea, erupción cutánea, edema localizado/hinchazón local y eritema) y vómito, durante el periodo de postcomercialización. Descripción de las reacciones adversas relevantes: Las reacciones adversas más notificadas fueron dos frecuentes (somnolencia y cefalea) y dos poco frecuentes (mareo y fatiga). Las frecuencias en bilastina frente a placebo fueron 3.06% vs. 2.86% para somnolencia; 4.01% vs. 3.38% para cefalea; 0.83% vs. 0.59% para mareo y 0.83% vs. 1.32% para fatiga. En casi todas las reacciones adversas mencionadas en la tabla anterior, se observó una incidencia similar en pacientes tratados con 20 mg de bilastina y en pacientes tratados con placebo. La información recogida durante la postcomercialización ha confirmado el perfil de seguridad observado durante el desarrollo clínico.
Interacciones medicamentosas y de otro genéro: Interacción con alimentos: Los alimentos reducen significativamente la biodisponibilidad oral de bilastina en un 30%. Interacción con jugo de toronja: La administración concomitante de bilastina 20 mg y jugo de toronja disminuyó la biodisponibilidad de bilastina en un 30%. Este efecto puede ocurrir también con otros jugos o derivados de las frutas. El grado de reducción de la biodisponibilidad puede variar entre fabricantes y frutos. El mecanismo responsable de esta interacción es la inhibición del OATP1A2, un transportador de captación, del cual bilastina es sustrato. Los medicamentos que sean sustratos o inhibidores del OATP1A2, tales como ritonavir o rifampicina, podrían igualmente reducir las concentraciones plasmáticas de bilastina. Interacción con ketoconazol o eritromicina: La administración concomitante de bilastina y ketoconazol o eritromicina aumentó el AUC de bilastina en 2 veces y la Cmax en 2-3 veces. Estos cambios se pueden explicar debido a la interacción con transportadores intestinales de excreción, ya que bilastina es sustrato de la P-gp y no es metabolizada. Estos cambios no parecen afectaral perfil deseguridadde bilastina yketoconazol oeritromicina, respectivamente. Otros medicamentos que sean sustratos o inhibidores de la de P-gp, tal como ciclosporina, podrían igualmente aumentar las concentraciones plasmáticas de bilastina. Interacción con diltiazem: La administración concomitante de bilastina 20 mg y diltiazem 60 mg aumentó la Cmax de bilastina en un 50%. Este efecto se puede explicar por la interacción con transportadores intestinales de excreción (Ver sección Farmacocinética y farmacodinamia) y no parece afectar al perfil de seguridad de bilastina. Interacción con alcohol: El rendimiento psicomotor después de la toma concomitante de alcohol y 20 mg de bilastina fue similar tras la administración de alcohol y placebo. Interacción con lorazepam: La administración concomitante de bilastina 20 mg y lorazepam 3 mg por 8 días no potenció los efectos depresores del SNC causados por lorazepam.
Alteraciones en los resultados de pruebas de laboratorio: Se realizaron evaluaciones en todos los estudios fases I, II y III tanto en la medición basal como durante y al concluir los estudios. No se observó ninguna desviación clínicamente ni estadísticamente signi