BLOTEX®
SANDOZ
Denominacion genérica: Atenolol.
Forma farmaceutica y formulación: Tabletas: Cada tableta contiene: Atenolol 50 mg y 100 mg. Excipiente 1 tableta
Indicaciones terapeuticas: Antihipertensivo (Betabloqueador): Hipertensión: tratamiento de la hipertensión. Puede usarse como agente único o concomitantemente con otros agentes antihipertensores, particularmente diuréticos tipo tiazídicos. Angina de pecho por arteriosclerosis coronaria: tratamiento a largo plazo de pacientes con angina de pecho. Infarto agudo del miocardio: tratamiento de pacientes hemodinamicamente estables con infarto agudo del miocardio definido o presumible, para reducir la mortalidad cardiovascular. El tratamiento puede iniciarse tan pronto como lo permita la condición clínica del paciente. (Ver: Dosis y vía de administracion y contraindicaciones). En general, no hay bases para tratar a pacientes como los excluidos del estudio ISIS-1 (presión arterial sistólica inferior a 100 mm Hg y frecuencia cardíaca inferior a 50 latidos por minuto), o los que tienen otras razones para evitar el betabloqueo. En algunos subgrupos, como los de pacientes de edad avanzada o aquéllos con presión arterial sistólica inferior a 120 mm Hg, los beneficios parecen menos probables.
Farmacocinética y farmacodinamia: Farmacocinética: el atenolol es un agente beta1-selectivo (cardioselectivo) bloqueador de los receptores betaadrenérgicos, sin actividad estabilizadora de membrana o simpaticomimética (agonista parcial) intrínseca. Sin embargo, este efecto preferencial no es absoluto, y en dosis altas el atenolol inhibe los beta2-adrenoceptores, principalmente los localizados en la musculatura bronquial y vascular. En humanos, la absorción de la dosis oral es rápida y consistente, aunque incompleta. Aproximadamente 50% de una dosis oral se absorbe en el tracto intestinal y el resto se excreta inalterada en las heces fecales. Los niveles sanguíneos máximos se alcanzan entre dos y cuatro horas después de la administración. A diferencia del propanolol o el metroprolol, y de manera similar al nadolol, el atenolol sufre poco o ningún metabolismo en el hígado, y la porción absorbida se elimina principalmente en la orina. Más del 85% de la dosis intravenosa se excreta en la orina, durante el curso de 24 horas, en comparación con el 50% de la dosis oral. El atenolol difiere del propanolol en que sólo una pequeña cantidad (6%-16%) se une a las proteínas plasmáticas. El perfil cinético produce niveles plasmáticos del fármaco relativamente consistentes, con aproximadamente una variación del cuádruple entre pacientes. La vida media de eliminación del atenolol oral es de aproximadamente 6-7 horas, y no hay alteración del perfil cinético del fármaco por la administración crónica. Después de la administración intravenosa los niveles plasmáticos máximos se alcanzan en el curso de 5 minutos. Los decrementos a partir de los niveles máximos son rápidos (entre 5 y 10 veces) durante las primeras 7 horas; a partir de entonces los niveles plasmáticos decrecen con una vida media similar a la del fármaco administrado oralmente. Después de dosis orales de 50 o 100 mg los efectos de betabloqueo y antihipertensores persisten al menos por 24 horas. Cuando hay insuficiencia renal, la eliminación de atenolol está estrechamente relacionada con la tasa de titulación glomerular; al descender la eliminación de creatinina por debajo de 35 ml/min/1,73 m2 existe acumulación significativa (ver Dosis y vía de administración). Farmacodinamia: en pruebas farmacológicas estándar hechas en animales o humanos, la actividad bloqueadora de los receptores betaadrenérgicos por parte del atenolol se ha demostrado por: 1) la reducción en la frecuencia cardíaca y el gasto cardíaco durante el reposo y el ejercicio, 2) la reducción de la presión arterial sistólica y diastólica durante el reposo y el ejercicio, 3) la inhibición de la taquicardia inducida por el isoproterenol, y 4) la reducción en la taquicardia ortostática refleja. En la primera hora siguiente a la administración oral de una dosis única de atenolol, resulta evidente un efecto significativo de betabloqueo, determinado por la reducción de la taquicardia durante el ejercicio. Este efecto alcanza su nivel máximo aproximadamente entre las 2 y las 4 horas, y se mantiene al menos por 24 horas. La máxima reducción de la taquicardia durante el ejercicio ocurre en los 5 minutos siguientes a la administración intravenosa. Con el fármaco administrado por vía oral e intravenosa la duración de la acción se relaciona con la dosis y también genera una relación lineal respecto a la concentración plasmática de atenolol. El efecto que ejerce en la taquicardia durante el ejercicio una dosis intravenosa de 10 mg se disipa considerablemente en 12 horas, mientras que la actividad betabloqueadora de dosis orales únicas de 50 y 100 mg sigue siendo evidente después de 24 horas de haberse administrado. Sin embargo, tal como se ha demostrado con todos los agentes betabloqueadores, el efecto antihipertensor no parece estar relacionado con el nivel plasmático. En sujetos normales, se ha demostrado la beta1-selectividad del atenolol, por su reducida capacidad para revertir el efecto vasodilatador del isoproterenol, mediado por los beta2-adrenoceptores, en comparación con dosis equivalentes betabloqueadoras de propanolol. En pacientes asmáticos una dosis de atenolol que produce un efecto mayor que el propanolol en la frecuencia cardíaca, provocó un incremento mucho menor en la resistencia de las vías aéreas. En un estudio comparativo de dosis orales equipotentes de varios betabloqueadores, controlado con placebo, el atenolol produjo un decremento significativamente inferior de FEV1 en comparación con los betabloqueadores no selectivos como el propanolol y, a diferencia de tales agentes, no inhibió la broncodilatación en respuesta al isoproterenol. En congruencia con este efecto cronotrópico debido al betabloqueo del nudo SA, el atenolol aumenta la longitud sinusal y el tiempo de recuperación del nudo sinusal. La conducción en el nudo AV también se prolonga. El atenolol carece de actividad estabilizadora de membrana, y al aumentar la dosis muy por encima de la que produce el betabloqueo no deprime la contractilidad del miocardio. Varios estudios han demostrado un incremento moderado (aproximadamente del 10%) en el volumen sistólico durante el reposo y el ejercicio. En estudios clínicos controlados, el atenolol administrado en una dosis oral diaria única fue un agente antihipertensor efectivo, al producir reducción de la presión arterial por 24 horas. Se ha estudiado el atenolol combinado con diuréticos tipo tiazídicos, y los efectos de la combinación en la presión arterial son aditivos. El atenolol también es compatible con la metildopa, la hidralazina y la prazosina, y cada una de estas combinaciones produce un decremento mayor de la presión arterial que el logrado con agentes únicos. El márgen de dosificación de atenolol es estrecho, y el incremento por encima de 100 mg administrados una vez diaria no está asociado con un efecto antihipertensor creciente. No se han establecido los mecanismos de los efectos antihipertensores de los agentes betabloqueadores. Se han propuesto varios mecanismos posibles, que incluyen: 1) el antagonismo competitivo de las catecolaminas en los lugares neuronales adrenérgicos periféricos (especialmente cardíacos), que conduce a la reducción del gasto cardíaco, 2) un efecto central que provoca la reducción del flujo simpático hacia la periferia, y 3) la supresión de la actividad de la renina. Los resultados de estudios de largo plazo no han demostrado ninguna disminución de la eficacia antihipertensora del atenolol con el uso prolongado. Mediante el bloqueo de los efectos positivos cronotrópicos e inotrópicos de las catecolaminas y la disminución de la presión arterial, el atenolol generalmente reduce los requerimientos de oxígeno en el corazón a cualquier nivel dado de esfuerzo, haciendo esto útil para muchos pacientes en el tratamiento a largo plazo de la angina de pecho. Por otra parte, el atenolol puede incrementar los requerimientos de oxígeno al aumentar la longitud de la fibra del ventrículo izquierdo y la presión diastólica final, particularmente en pacientes con insuficiencia cardíaca. En un estudio clínico multicéntrico (ISIS-1) realizado en 16.027 pacientes con presunto infarto del miocardio, los pacientes que se presentaban en el curso de 12 horas (media = 5 horas) a partir de la manifestación del dolor fueron asignados aleatoriamente a terapia convencional más atenolol (n = 8.037) o sólo a terapia convencional (n = 7.990). Se excluyó a pacientes con frecuencia cardíaca inferior a 50 latidos por minuto o presión arterial sistólica inferior a 100 mm Hg, o con otras contraindicaciones para el betabloqueo. El 38% de cada grupo fue tratado en las 4 horas siguientes a la manifestación del dolor. El tiempo medio desde la manifestación del dolor hasta la admisión fue de 5,0 ± 2,7 horas en ambos grupos. Los pacientes del grupo de atenolol habrían de recibir una inyección de 5 a 10 mg, aplicada en el curso de 5 minutos, más tabletas de 50 mg de atenolol administradas cada 12 horas en el primer día del estudio (primera dosis oral administrada aproximadamente 15 minutos después de la dosis intravenosa), y después una tableta diaria de 100 mg de atenolol o dos tabletas diarias de 50 mg de atenolol durante los días 2 a 7. Los grupos eran similares en cuanto a las características demográficas y de historia clínica, así como en la evidencia electrocardiográfica de infarto del miocardio, bloqueo de rama y bloqueo auriculoventricular de primer grado en el momento de la admisión. Durante el período de tratamiento (días 0 a 7) las tasas de mortalidad vascular fueron de 3,89% en el grupo de atenolol (313 muertes) y de 4,57% en el grupo de control (365 muertes). Esta diferencia absoluta de 0,68% en las tasas es estadísticamente significativa en el nivel de P < 0,05. La diferencia absoluta se traduce en una reducción proporcional de 15% (3,89 a 4,57/4,57 = -0,15). Los límites de confianza de 95% son de 1-27%. En su mayor parte, la diferencia se atribuyó a la mortalidad de los días 0 a 1 (atenolol - 121 muertes; control - 171 muertes). A pesar del tamaño considerable del estudio ISIS-1, no es posible identificar claramente a subgrupos de pacientes con la mayor o la menor probabilidad de beneficiarse por el tratamiento temprano con atenolol. Sin embargo, el buen juicio clínico sugiere que los pacientes que dependen de la estimulación simpática para mantener un gasto cardíaco y una presión arterial adecuados, no son buenos candidatos para el betabloqueo. De hecho, el protocolo del estudio reflejó ese juicio al excluir a pacientes con presión arterial sistólica consistentemente inferior a 100 mm Hg. Los resultados generales del estudio son compatibles con la posibilidad de que los pacientes con presión arterial limítrofe (inferior a 120 mm Hg de presión sistólica), especialmente los mayores de 60 años, tengan menos probabilidades de beneficiarse. Se desconoce el mecanismo mediante el cual el atenolol mejora la supervivencia en pacientes con infarto agudo del miocardio, definido o presunto, como es el caso de otros betabloqueadores en la condición posterior al infarto. Además de sus efectos en la supervivencia, se han demostrado otros beneficios clínicos del atenolol que incluyen la reducción de la frecuencia de los latidos ventriculares prematuros, la disminución del dolor de pecho y una menor elevación enzimática.
Contraindicaciones: Las tabletas de atenolol están contraindicadas en la bradicardia sinusal, choque cardiogénico e insuficiencia cardíaca manifiesta (ver Precauciones).
Precauciones generales: Advertencias: insuficiencia cardíaca: es necesaria la estimulación simpática para soportar la función circulatoria en la insuficiencia cardíaca congestiva, y el betabloqueo conlleva el riesgo potencial de deprimir la contractilidad del miocardio y agudizar la insuficiencia. En pacientes con insuficiencia cardíaca congestiva controlados con digital y/o diuréticos debe administrarse con precaución el BLOTEX®, pues ambos agentes reducen la conducción auriculoventricular. El tratamiento con betabloqueadores está contraindicado en pacientes con infarto agudo del miocardio no controlados pronta y eficazmente con 80 mg de furosemida intravenosa o una terapia equivalente. Pacientes sin antecedentes de insuficiencia cardíaca: en algunos casos, la depresión continua del miocardio con agentes betabloqueadores durante un tiempo puede provocar insuficiencia cardíaca. Al primer signo o síntoma de insuficiencia cardíaca inminente, debe digitalizarse totalmente a los pacientes y/o administrárseles un diurético, así como observarse la respuesta muy de cerca. Si persiste la insuficiencia cardíaca a pesar de la digitalización y la diuresis adecuadas, debe suspenderse el BLOTEX®. (Ver Dosis y vía de administración). Suspensión de la terapia con BLOTEX®: a los pacientes con enfermedad coronaria que estén siendo tratados con BLOTEX® debe advertírseles que no deben suspender abruptamente la terapia. Se ha reportado exacerbación severa de angina, así como infarto del miocardio y arritmias ventriculares, después de la suspensión abrupta de la terapia con betabloqueadores. Las dos últimas complicaciones pueden ocurrir con sin exacerbación previa de la angina de pecho. Igual que con otros betabloqueadores, cuando se planee suspender el BLOTEX® debe observarse cuidadosamente a los pacientes y pedírseles que limiten al mínimo la actividad física. Si empeora la angina de pecho o se desarrolla insuficiencia coronaria aguda, se recomienda reinstituir prontamente el BLOTEX®, al menos temporalmente. Debido a que la enfermedad coronaria es común y puede no reconocerse, podría ser prudente no suspender abruptamente la terapia con BLOTEX® incluso cuando se trate a los pacientes sólo por hipertensión (ver Dosis y vía de administración). Uso concomitante de bloqueadores de canales del calcio: puede ocurrir bradicardia y bloqueo cardíaco, así como aumentar la presión diastólica final del ventrículo izquierdo, cuando se administran betabloqueadores con verapramil o diltiazem. Los pacientes con anormalidades preexistentes en la conducción o disfunción ventricular izquierda son particularmente susceptibles. Enfermedades broncospásticas: los pacientes con enfermedad broncospastica, en general, no deben recibir betabloqueadores. Sin embargo, debido a su beta1-selectividad, el BLOTEX® puede usarse con precaución en pacientes con enfermedad broncospástica que no respondan a otro tratamiento antihipertensor o no puedan tolerarlo. Dado que la beta1-selectividad no es absoluta debe usarse la dosis más baja posible de BLOTEX®, iniciando la terapia con 50 mg, y debe disponerse de un agente beta2-estimulante (broncodilatador). Si es preciso aumentar la dosificación, debe considerarse el dividir la dosis para alcanzar niveles sanguíneos máximos menores. Anestesia y cirugía mayor: no es recomendable suspender los fármacos bloqueadores de los betaadrenoceptores antes de la cirugía, en la mayoría de los pacientes. Sin embargo, debe tenerse cuidado al usar anestésicos como los que pueden deprimir el miocardio. Si ocurre dominancia vagal, puede corregirse con atropina (1 a 2 mg por vía intravenosa). El BLOTEX®, igual que otros betabloqueadores, es un inhibidor competitivo de los agonistas de los betarreceptores y sus efectos en el corazón pueden revertirse administrando con precaución tales agentes, como la dobutamina o el isoproterenol (Ver Manifestaciones y manejo de la sobredosificación o ingesta accidental). Diabetes e hipoglucemia: el BLOTEX® debe usarse con precaución en pacientes diabéticos que requieran un agente betabloqueador. Los betabloqueadores pueden enmascarar la taquicardia que ocurre con la hipoglucemia, pero otras manifestaciones como el mareo y la sudoración puede no resultar afectadas significativamente. En las dosis recomendadas, el BLOTEX® no potencia la hipoglucemia inducida por la insulina y, a diferencia de los betabloqueadores no selectivos, no retrasa el restablecimiento de los niveles sanguíneos normales de glucosa. Tirotoxicosis: el bloqueo betadrenérgico puede enmascarar ciertos signos clínicos del hipertiroidismo (como la taquicardia). La suspensión abrupta del betabloqueo podría precipitar una tempestad tiroidea; por tanto, debe vigilarse muy de cerca a los pacientes con presunta tirotoxicosis a quienes se les suspenda la terapia con BLOTEX®.
Restricciones de uso durante el embarazo y la lactancia: El BLOTEX® puede causar daño fetal en mujeres embarazadas. El fármaco atraviesa la barrera placentaria y aparece en la sangre del cordón umbilical. La administración de BLOTEX® a partir del segundo trimestre de embarazo se ha asociado con el nacimiento de infantes de menor tamaño que el correspondiente a la edad de gestación. No se han realizado estudios sobre el uso de BLOTEX® en el primer trimestre de embarazo, y no puede excluirse la posibilidad de daño fetal. Si se usa el fármaco durante el embarazo, o si la paciente se embaraza durante el tratamiento con BLOTEX® debe informársele el riesgo potencial para el feto. Se ha demostrado que el BLOTEX® produce en la rata un incremento, relacionado con la dosis, en las resorciones embrio-fetales al usar dosis diarias de 50 mg/kg o superiores, o 25 veces o más la dosis antihipertensora máxima recomendada para humanos.*Aunque no se observaron efectos similares en conejos, no se evaluó el compuesto administrado en dosis diarias superiores a 25 mg/kg o 12.5 veces la dosis antihipertensora máxima recomendada para humanos. *Con base en la dosis máxima de 100 mg diarios para un paciente de 50 kg. Lactancia: el BLOTEX® se excreta a través de la leche humana en una relación de 1,5 a 6,8 con respecto a la concentración plasmática. Debe tenerse precaución al administrar BLOTEX® a las madres durante la lactancia. Se ha reportado bradicardia clínicamente significativa en infantes lactantes. Los infantes prematuros o con insuficiencia renal son más propensos a experimentar efectos adversos. Los pacientes que ya estén siendo tratados con un betabloqueador deben ser evaluados cuidadosamente antes de administrarles BLOTEX® Las dosis inicial y subsecuentes de BLOTEX® pueden ajustarse para reducirlas conforme a las observaciones clínicas, que incluyen mediciones de la frecuencia cardíaca y de la presión arterial. El BLOTEX® puede agravar los trastornos circulatorios arteriales periféricos.
Reacciones secundarias y adversas: La mayoría de los efectos adversos han sido leves y transitorios. Las frecuencias estimadas de la tabla siguiente se derivaron de estudios controlados en pacientes hipertensos en los cuales los eventos adversos se reportaron voluntariamente (estudios de E.U.A.) o recabándola; p. ej., mediante listas de verificación (estudios de otros países). La frecuencia de los efectos adversos recabados fue mayor para pacientes tratados con BLOTEX® y con placebo que los efectos reportados voluntariamente. La relación causal es incierta cuando es similar la frecuencia de los efectos adversos del BLOTEX® y del placebo.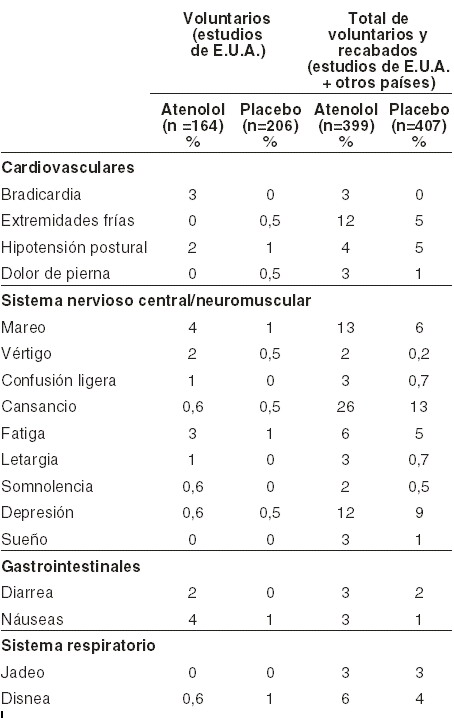
Infarto agudo del miocardio: en una serie de investigaciones para el tratamiento del infarto agudo del miocardio, como era de esperarse con cualquier betabloqueador, la bradicardia y la hipotensión ocurrieron más comúnmente en pacientes tratados con BLOTEX® que en los pacientes de control. Sin embargo, generalmente respondieron a la atropina y/o la suspensión de la administración de BLOTEX®. La incidencia de insuficiencia cardíaca no aumentó con el BLOTEX®. A menudo se usaron agentes inotrópicos. La frecuencia reportada de estos y otros eventos ocurridos durante estas investigaciones se indica en la tabla siguiente. En un estudio con 477 pacientes se reportaron estos eventos adversos durante la administración oral y/o intravenosa de atenolol: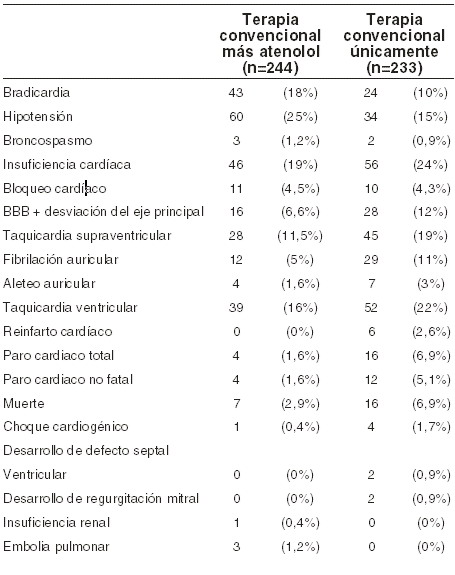
En el Estudio Internacional de Supervivencia al Infarto (ISIS-1) que incluyó a más de 16.000 pacientes, 8.037 de los cuales fueron asignados aleatoriamente para que recibieran tratamiento con BLOTEX®, la dosificación intravenosa y la subsecuente administración oral de BLOTEX® se interrumpieron o redujeron por las siguientes razones: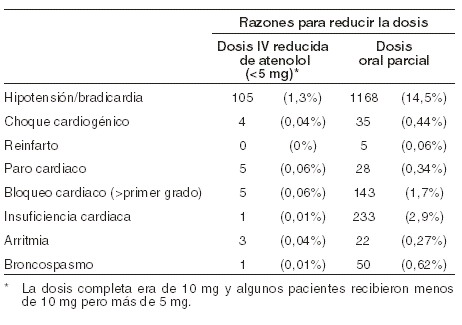
Durante la experiencia con BLOTEX® posterior a la comercialización se ha reportado lo siguiente, que se relaciona con el uso temporal del fármaco: aumento de enzimas hepáticas y/o bilirrubina, alucinaciones, cefalea, impotencia, enfermedad de Peyronie, hipotensión postural que puede asociarse con síncope, exantema psoriasiforme o exacerbación de psoriasis, psicosis, púrpura, alopecia reversible, trombocitopenia y trastornos visuales. Igual que otros betabloqueadores, al BLOTEX® se le ha asociado con el desarrollo de anticuerpos antinúcleo y síndrome de lupus. Efectos adversos potenciales: además se ha reportado una diversidad de efectos adversos con otros agentes bloqueadores betaadrenérgicos, y pueden considerarse efectos adversos del BLOTEX®. Hematologicos: agranulocitosis. Alergicos: fiebre, combinada con dolor e irritación de garganta, laringospasmo y dificultad para respirar. Sistema nervioso central: depresión reversible que evoluciona en catatonia, síndrome agudo reversible caracterizado por desorientación en cuanto a tiempo y lugar, pérdida de memoria de corto plazo, susceptibilidad emocional con obnubilación ligera, y declinación del rendimiento neuropsicométrico. Gastrointestinales: trombosis arterial mesentérica y colitis isquémica. Otros: exantema eritematoso, fenómeno de Raynaud. Diversos: se han reportado erupciones cutáneas y/o resequedad en los ojos, asociados con el uso de fármacos bloqueadores betaadrenérgicos. La incidencia reportada es reducida y, en la mayoría de los casos, los síntomas han desaparecido al suspender el tratamiento. Debe considerarse la suspensión del fármaco si cualquiera de las reacciones no es explicable. Debe vigilarse muy de cerca de los pacientes después de suspender la terapia (ver Dosis y vía de administración). El síndrome oculomucocutáneo asociado con el betabloqueador practolol no se ha reportado con el BLOTEX®. Aún más, numerosos pacientes que habían experimentado reacciones establecidas al practolol fueron transferidos a la terapia con BLOTEX®, con la subsecuente resolución o quiescencia de las reacciones.
Interacciones medicamentosas y de otro género: Los fármacos depletores de las catecolaminas (como la reserpina) pueden tener un efecto aditivo cuando se administran con agentes betabloqueadores. Por tanto, debe observarse muy de cerca a los pacientes tratados con BLOTEX® más un depletor de catecolaminas, para detectar evidencias de hipotensión y/o bradicardia marcada, que puede producir vértigo, síncope o hipotensión postural. Los bloqueadores de canales del calcio también pueden tener un efecto aditivo cuando se administran con BLOTEX® Los betabloqueadores pueden exacerbar la hipertensión de rebote que podría seguir a la suspensión de clonidina. Si se administran concomitantemente ambos fármacos, el betabloqueador debe suspenderse varios días antes de la interrupción gradual de la clonidina. Si la terapia con betabloqueadores sustituye a la clonidina, la introducción de los betabloqueadores debe iniciarse varios días después de suspender la administración de clonidina. La información sobre el uso concomitante de BLOTEX® y aspirina es limitada. Los datos de varios estudios, por ejemplo TIMI-II e ISIS-2, no sugieren actualmente ninguna interacción clínica entre la aspirina y los betabloqueadores en la manifestación del infarto agudo del miocardio. Durante el tratamiento con betabloqueadores, los pacientes con antecedentes de reacción anafiláctica a una diversidad de alergenos pueden tener una reacción más severa por la exposición repetida, ya sea accidental, diagnóstica o terapéutica. Tales pacientes pueden no responder a las dosis habituales de adrenalina usadas para tratar la reacción alérgica.
Alteraciones en los resultados de pruebas de laboratorio: En algunas ocasiones se ha observado incremento en enzimas hepáticas.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Dos estudios de largo plazo en ratas (con duración máxima de dosificación de 18 o 24 meses) y otros de largo plazo en ratones (cuya duración máxima de dosificación fue de 18 meses), en cada uno de los cuales se usaron niveles de dosis de hasta 300 mg/kg diarios o 150 veces la dosis máxima antihipertensora recomendada en humanos,* no produjeron indicios de potencial carcinogénico del BLOTEX® En un tercer estudio (de 24 meses) en ratas, con dosis de 500 y 1.500 mg/kg diarios (250 y 750 veces la dosis máxima antihipertensora recomendada en humanos*) produjo mayores incidencias de tumores medulares adrenales benignos en machos y hembras, fibroadenomas mamarios en las hembras, así como adenomas pituitarios anteriores y carcinomas de las células parafoliculares tiroideas en los machos. No hubo evidencia de potencial mutagénico del BLOTEX® en la prueba letal dominante (en ratones), en la prueba de citogenética in vivo (criceto chino) o la prueba de Ames (S typhimurium). La administración de BLOTEX® no afectó la fertilidad en ratas machos ni hembras (evaluados con niveles de dosificación de hasta 200 mg/kg diarios o 100 veces la dosis máxima recomendada para humanos*). Toxicología en animales: estudios crónicos en animales, con el uso de BLOTEX® oral, revelaron que ocurrió vacuolación en las células epiteliales de las glándulas de Brunner del duodeno en perros machos y hembras, con todos los niveles de dosificación de BLOTEX® probados (empezando por 15 mg/kg diarios o 7,5 veces la dosis antihipertensora máxima recomendada para humanos*), y que aumentó la incidencia de degeneración auricular en ratas machos con 300 mg/kg diarios de BLOTEX® más no con 150 mg/kg diarios (respectivamente 150 y 75 veces la dosis antihipertensora máxima recomendada para humanos*). *Con base en la dosis máxima de 100 mg diarios para un paciente de 50 kg. Embarazo: efectos teratogénicos, embarazo categoría D (ver Precauciones o restricciones de uso durante el embarazo y la lactancia).
Dosis y vía de administración:: Oral. Hipertensión: La dosis inicial de BLOTEX® es de 50 mg, administrados en una tableta diaria, sola o añadida a una terapia diurética. El efecto total de esta dosis se observará generalmente en el curso de una a dos semanas. Si no se logra una respuesta óptima, debe aumentarse la dosis a 100 mg de BLOTEX® en una tableta diaria. Es improbable que el incremento de la dosificación por encima de 100 mg diarios produzca mayor beneficio. El BLOTEX® debe usarse solo o concomitantemente con otros agentes antihipertensores, incluyendo los diuréticos tipo tiazídicos, la hidralazina, la prazosina y la alfametildopa. Angina de pecho: la dosis inicial de BLOTEX® es de 50 mg, administrados en una tableta diaria. Si no se logra una respuesta óptima, debe aumentarse la dosis a 100 mg de BLOTEX® en una tableta diaria. Algunos pacientes pueden requerir la administración única de 200 mg diarios para lograr el efecto óptimo. El control durante 24 horas con una dosis única diaria se logra administrando dosis superiores a la necesaria para alcanzar un efecto máximo inmediato. El efecto temprano máximo en la tolerancia al ejercicio ocurre con dosis de 50 a 100 mg, pero con tales dosis el efecto a las 24 horas se atenúa, promediando aproximadamente entre 50 y 75% del observado con la administración oral única de 200 mg diarios. Al administrarse BLOTEX® oralmente en una dosis única diaria de 100 mg o dos dosis diarias de 50 mg, por 6 a 9 días o hasta que el paciente sea dado de alta en el hospital. Si se experimenta bradicardia o hipotensión que requiera tratamiento, o cualquier otro efecto adverso, debe suspenderse el BLOTEX® Los datos de estudios efectuados con otros betabloqueadores sugieren que si existe duda en cuanto al uso de betabloqueadores intravenosos o a la estimación clínica de que exista contraindicación, puede eliminarse el betabloqueador intravenoso y a los pacientes que cumplan con los criterios de seguridad pueden administrárseles dos tabletas diarias de 50 mg de BLOTEX® o una tableta diaria de 100 mg, al menos durante siete días (si se excluye la administración intravenosa). Si bien la demostración de la eficacia del BLOTEX® se basa totalmente en datos referentes a los primeros siete días siguientes al infarto, los datos obtenidos en estudios de otros betabloqueadores sugieren que el tratamiento con betabloqueadores que son efectivos inmediatamente después del infarto puede continuarse durante uno a tres años, si no hay contraindicaciones. El uso de BLOTEX® es adicional a la terapia estándar en la unidad de coronarias. Pacientes de edad avanzada con insuficiencia renal: el BLOTEX® se excreta por vía renal, por lo que la dosis debe ajustarse en casos de insuficiencia renal severa. Para pacientes de edad avanzada también puede ser apropiada cierta reducción en la dosis, puesto que la reducción de la función renal es una consecuencia fisiológica del envejecimiento. Se esperaría que la excreción de BLOTEX® decrezca con la edad. No ocurre acumulación significativa de BLOTEX® sino hasta que la eliminación de creatinina es inferior a 35 ml/min/1,73 m2. La acumulación de atenolol y la prolongación de su vida media se estudiaron en sujetos con eliminación de creatinina entre 5 y 105 ml/min. Los niveles plasmáticos máximos aumentaron significativamente con eliminaciones de creatinina inferiores a 30 ml/min. Se recomiendan las siguientes dosificaciones orales máximas para pacientes de edad avanzada con insuficiencia renal y para aquéllos con insuficiencia renal debida a otras causas:
Algunos pacientes con insuficiencia renal o de edad avanzada tratados por hipertensión pueden requerir una dosis inicial inferior de BLOTEX®: 25 mg en una tableta diaria. Si se usa esta dosis de 25 mg la eficacia debe evaluarse cuidadosamente. Esto debe incluir la medición de la presión arterial antes de la siguiente dosis (presión arterial deprimida), para comprobar que el efecto persiste durante 24 horas. Aunque puede considerarse una reducción similar de la dosis para pacientes de edad avanzada y/o con insuficiencia renal que estén siendo tratados para otras indicaciones distintas de la hipertensión, no hay datos disponibles sobre tales poblaciones. Los pacientes sometidos a hemodiálisis deben recibir 25 o 50 mg de BLOTEX® después de cada diálisis, bajo supervisión hospitalaria pues pueden ocurrir descensos acentuados de la presión arterial. Suspensión de la terapia en pacientes con angina de pecho: si se planea suspender la terapia con BLOTEX®, debe hacerse gradualmente y los pacientes deben ser observados cuidadosamente, así como pedírseles que limiten al mínimo la actividad física. Uso pediátrico: no se han establecido la seguridad y la efectividad en pacientes pediátricos.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: Se ha reportado sobredosificación en pacientes que sobreviven a dosis agudas de hasta 5 g de BLOTEX®. Se ha reportado la muerte de un hombre que pudo haber ingerido hasta 10 g. Los síntomas predominantes reportados después de las sobredosificaciones de BLOTEX® son letargia, trastornos respiratorios, jadeo, pausa sinusal y bradicardia. Asimismo, los efectos comunes asociados con la sobredosificación de cualquier agente bloqueador betaadrenérgico y que podrían esperarse por sobredosis de BLOTEX® incluyen: insuficiencia cardíaca congestiva, hipotensión, broncospasmo y/o hipoglucemia. El tratamiento de la sobredosificación debe orientarse a la eliminación del fármaco no absorbido, mediante vómito inducido, lavado estomacal o administración de carbón activado. El BLOTEX® puede eliminarse de la circulación general mediante hemodiálisis. A criterio del médico deben usarse otras modalidades de tratamiento, que pueden incluir: bradicardia: atropina intravenosa. Si no hay respuesta al bloqueo vagal, puede administrarse isoproterenol cuidadosamente. En casos refractarios puede resultar indicado un marcapaso cardíaco transvenoso. Bloqueo cardíaco (de segundo o tercer grado): isoproterenol o marcapaso cardíaco transvenoso. Insuficiencia cardíaca: digitalizar al paciente y administrar un diurético. Se ha reportado la utilidad del glucagón. Hipotensión: vasopresores como la dopamina o la norepinefrina. Vigilar la presión arterial continuamente. Broncospasmo: un beta2-estimulante como el isoproterenol o la terbutalina y/o aminofilina. Hipoglucemia: glucosa intravenosa. Según la gravedad de los síntomas, puede requerirse cuidado intensivo e instalaciones para brindar apoyo cardíaco y respiratorio.
Presentación(es): Caja con 14 y 28 tabletas de 50 mg y 100 mg para Venta al Público.
Recomendaciones sobre almacenamiento: Consérvese a temperatura ambiente a no más de 30°C y en lugar seco.
Leyendas de protección: Su venta requiere receta médica. No se deje al alcance de los niños. Literatura exclusiva para médicos.
Nombre y domicilio del laboratorio: Hecho en EUA por: Sandoz Inc.2555 West Midway Bouylevard, P.O. Box 446, Broomfield, Colorado 80038-0446, EUA. Acondicionado y distribuido por: Novartis Farmacéutica, S.A. de C.V., Calz. de Tlalpan 1779, Col. San Diego Churubusco, C.Cp. 04120 México, D.F.
Número de registro del medicamento: 424M99 SSA IV
Clave de IPPA: LEAR-05330020510582/RM2005