BONVIVA®
MOKSHA8
Tabletas
Denominación genérica: Acido lbandronico
Forma farmacéutica y formulación: Tableta. Cada tableta contiene: lbandronato de sodio monohidratado equivalente a de acido ibandronico Excipiente c.b.p
Indicaciones terapéuticas: Tratamiento de la osteoporosis posmenopausica: 150 mg 1 tableta Bonviva® 150 mg esta indicado para el tratamiento de la osteoporosis posmenopausica, para reducir el riesgo de fracturas. La osteoporosis puede confirmarse por los valores de masa ósea (puntuacion T: < -2.0 DE) y/o por la presencia o antecedentes de fracturas osteoporoticas, o una masa ósea baja (T: < -2.5 DE) en ausencia de antecedentes documentados de fracturas osteoporoticas pre-existentes. Prevencion de la osteoporosis posmenopausica: Bonviva® 150 mg esta indicado para la prevenci6n de la osteoporosis en mujeres posmenopausicas que todavia no presentan osteoporosis, pero que estan en mayor riesgo de presentar fracturas segun los criterios de las guias clinicas actuales. Limitaciones de uso importantes No se ha determinado el tiempo de uso optimo. Debe considerarse la interrupcion del farmaco despues de 3 a 5 años de uso en las pacientes con un riesgo bajo de presentar fracturas (es decir, tratadas para la prevencion de la osteoporosis). A las pacientes a quienes se les interrumpa la terapia deberan realizarse una evaluacion periodica de su riesgo de presentar fracturas.
Farmacocinética y farmacodinamia: Farmacodinamia. La accion farmacodinamica del acido ibandronico es la inhibicion de la resorcion ósea. In vivo, el acido ibandronico previene la destruccion ósea inducida experimentalmente por bloqueo de la funcion gonadal, retinoides, tumores o extractos tumorales. En ratas jovenes (de rapido crecimiento), la resorcion ósea endogena se inhibe tambien, conduciendo al incremento de la masa ósea comparada con la de los animales sin tratamiento. Los modelos animales confirman que el acido ibandronico es un potente inhibidor de la actividad osteoclastica. En ratas en crecimiento, no existe evidencia de mineralizacion deteriorada aunque fueron tratadas con dosis mayores a 5,000 veces superiores de la requerida para el tratamiento de la osteoporosis. La gran potencia y el gran margen terapeutico del acido ibandronico permiten mayor flexibilidad de las pautas posologicas asi como la posibilidad de instaurar un tratamiento intermitente con dosis relativamente bajas e intervalos prolongados sin medicacion. La administraci6n prolongada - diaria o intermitente de acido ibandronico (con intervalos prolongados libres de dosis) a ratas, perros y monos se asocio a la formacion de tejido óseo nuevo de calidad normal y/o a un aumento de la resistencia mecanica incluso con dosis superiores a las farmacologicamente previstas, tambien dentro del intervalo toxico. Con el estudio clinico MF4411 en humanos, se confirmo la eficacia anti-fractura de la administraci6n diaria o mensual de Bonviva® con un intervalo de 9 - 10 semanas libre de dosis de acido ibandronico. Tanto la administraci6n diaria como intermitente (con un intervalo de 9-10 semanas sin medicacion por trimestre) de Bonviva® por via oral en las mujeres posmenopausicas produjo cambios bioquimicos indicativos de la inhibicion (dependiente de la dosis) de la resorcion ósea, como supresipn de marcadores bioquimicos urinarios de la degradacion del colageno óseo (p.ej.: desoxipiridinolina y telopeptidos C y N entrecruzados de colageno tipo I. Tras suspender el tratamiento, hay una reversion a los indices patologicos pre-terapeuticos de elevacion de la resorcion osea caracteristicos de la osteoporosis posmenopausica. El analisis histologico de las biopsias óseas tomadas a los dos y tres afios de tratamiento en mujeres posmenopausicas ha puesto de manifiesto un tejido óseo de calidad normal, sin signos de mineralización defectuosa. En un estudio de bioequivalencia Fase 1 realizado en 72 mujeres posmenopausicas que recibiran oralmente 150 mg cada 28 dias para un total de cuatro dosis, se observo inhibicion del CTX serico despues de la primera dosis a las 24 horas con una inhibiciónmedia máxima del 69% observada a la tercera y cuarta dosis., inhibici6n media maxima despues fue del 740/o con una reduccion en la inhibicion media del 560/o observada 28 dias despues de la cuarta dosis. Sin mas administraciones, ya no hay supresión de los marcadores de resorcion ósea. Mecanismo de accion El acido ibandrónico es un potente bifosfonato nitrogenado, que actua en el tejido óseo y especfficamente inhibe la actividad de los osteoclastos, sin interferir con el reclutamiento osteoclastico. La accion selectiva del acido ibandronico sobre el tejido óseo se debe a su alta afinidad por la hidroxiapatita, la cual constituye la matriz mineral ósea. El acido ibandronico reduce la resorcion ósea, sin tener efecto directo sobre la formaci6n de los huesos. En las mujeres posmenopausicas, disminuye el recambio óseo acelerado hacia niveles premenopausicos, con lo que se produce una ganancia neta progresiva de masa ósea. La administracion diaria o intermitente del acido ibandronico disminuye la resorcion ósea, dada por la disminuci6n de las concentraciones sericas y urinarias de los marcadores bioqufmicos de recambio óseo, el aumento de la OMO y la menor incidencia de fracturas. Eficacia (estudios clinicos) Tratamiento de la osteoporosis posmenopausica En un estudio clfnico inicial anti-fractura (MF 4411) doble ciego, aleatorizado controlado con placebo, de tres arios de duración, se demostro una disminucion estadisticamente significativa y relevante desde el punto de vista medico, en la incidencia de nuevas fracturas vertebrales radiograficas, morfometricas y clfnicas. Bonviva® fue evaluado en dosis orales de 2.5 mg diarios y 20 mg de forma intermitente (20 mg en dias alternos hasta completar 12 dosis al comienzo de cada ciclo de tres meses, separados por periodos de reposo farmacol6gico de 9-10 semanas). Bonviva® se administr6 60 minutos antes del primer alimento o bebida del dia (perfodo de ayuno posterior a la dosis). En el estudio participaron 2,946 mujeres de 55 a 80 años (de ellas 2,928 fueron incluidas para el analisis de eficacia), con una OMO de 2 a 5 desviaciones estandar (DE) lumbares por debajo del fndice premenopausico media (puntuacion en al menos en una vertebra lumbar (L 1-L4) y antecedentes de una a cuatro fracturas vertebrales previas. Todas las pacientes recibieron 500 mg de calcio y 400 UI de vitamina D al dia. Para ambos grupos de tratamiento con Bonviva® se demostro una reducci6n - estadfsticamente significativa y relevante desde el punto de vista medico - en la incidencia de nuevas fracturas vertebrales. La incidencia de nuevas fracturas vertebrales radiograficas disminuyo en un 620/o durante los tres dias del estudio con el regimen de 2.5 mg diarios. Las fracturas vertebrales clinicas tambien disminuyeron en un 490/o. Este potente efecto sabre las fracturas verbales, se confirmo con una disminucion estadisticamente significative de la reduccion en la estatura en comparacion con el placebo. Este efecto de protecci6n frente a las fracturas fue constante durante todo el estudio. No se apreciaron indicios de desvanecimiento del efecto con el tiempo. Aunque este estudio clfnico no estaba especificamente diseñado para demostrar la eficacia del acido ibandronico en fracturas extravertebrales, en un subgrupo de pacientes de alto riesgo de fractura, (OMO cervicofemoral T < -3.0 DE) pudo demostrarse una disminuci6n del riesgo relativo de fracturas extravertebrales de magnitud semejante a la observada para las fracturas vertebrales (69%). La observacion de la eficacia en relacion con las fracturas extravertebrales en subgrupos de alto riesgo concuerda bien con las resultados de estudios clfnicos para otros bifosfonatos. Con el regimen de administraci6n diaria, la OMO lumbar aumento un 5.30/o con respecto al placebo al cabo de 3 años de estudio. En comparaci6n con los valores iniciales, el aumento fue de 6.5%. Los marcadores bioquimicos del recambio óseo (tales coma CTX urinario y osteocalcina serica) demostraron el patron previsto de inhibicion hasta niveles premenopausicos con valores maximos de inhibici6n al cabo de 3-6 meses. Tan solo un mes despues del inicio del tratamiento con Bonviva® se observo una reduccion del 50% y 78% (ibandronato diario e intermitente respectivamente) en las marcadores bioqufmicos de la resorcion ósea, en ambos casos de trascendencia clinica. La disminucion de las marcadores bioqufmicos de la resorcion ósea fue evidente desde la primera semana del tratamiento. Bonviva® 150 mg una vez al mes Densidad mineral ósea (OMO) Bonviva® 150 mg una vez al mes, mostro ser tan eficaz coma la dosis diaria de Bonviva® 2.5 mg en un estudio multicentrico doble ciego a dos años, (BM 16549) de mujeres postmenopausicas con osteoporosis (T OMO inicial de la columna lumbar por debajo de -2.5 DE). Esto ha sido demostrado tanto en el analisis primario a un año coma en el analisis confirmatorio a dos años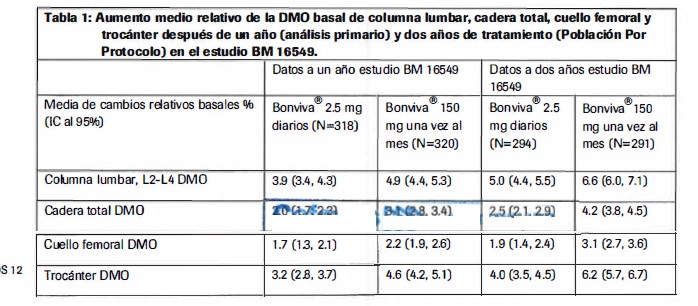
Ademas, Bonviva® 150 mg, una vez al mes fue superior a la dosis diaria de Bonviva® 2.5 mg para incrementos en la OMO de la columna lumbar de acuerdo al analisis prospectivo a un año, p=0.002 ya dos años, p < 0.001. Al primer año (analisis primario), 91.3% (p=0.005) de las pacientes que recibieron Bonviva® 150 mg una vez al mes presentaron un incremento en la DMO lumbar de columna por arriba o igual al basal (Respondedores OMO) comparado con un 84% de pacientes con tratamiento diario de Bonviva® 2.5 mg. A los dos años 93.5% (p=0.004) y 86.4% de las pacientes con Bonviva® 150 mg una vez al mes o Bonviva® 2.5 % mg diarios, respectivamente fueron respondedores.. Marcadores bioquimicos del recambio óseo Se observaron reducciones clinicamente significativas en todas las mediciones de niveles sericos de CTX para los meses 3, 6, 12 y 24. Despues de un año (analisis primario), la mediana de los cambios relativos del valor basal era -76% para Bonviva® 150 mg una vez al mes y -67% para Bonviva® 2.5 mg diarios. A los dos afios el cambio medio relativo fue -68% y- 62% en el brazo de 150 mg mensuales y 2.5 mg diarios, respectivamente. Al primer año, 83.5% (p=0.006) de las pacientes tratados con Bonviva® 150 mg una vez al mes y 73.9% de las pacientes tratados con Bonviva® 2.5 mg diarios, fueron identificados como respondedoras (definidas como un descenso de > 50% del valor basal). A los dos años 78.7% (p=0.002) y 65.5% de las pacientes fueron identificadas como respondedoras en los grupos de 150 mg mensuales y 2.5 mg diarios respectivamente. Considerando los resultados del estudio BMl 6549, se espera que Bonviva 150 mg una vez al mes, sea al menos mas eficas como la dosis diaria. Prevencion de la osteoporosis posmenopausica La prevencion de la perdida ósea se demostro en un estudio clinico (MF 4499) de diseño doble ciego y comparativo con placebo con duracion de dos años con el cambio de la OMO vertebral como criterio principal de eficacia. En este estudio se compararon 3 niveles de dosis del acido ibandronico en administracion diaria (0.5 mg, 1.0 mg, 2.5 mg) con placebo. Cada paciente recibio un suplemento de 500 mg diarios de calcio. En el estudio participaron 653 mujeres posmenopausicas sin osteoporosis estratificadas (de ellas, 648 incluidas en el analisis de eficacia) segun el tiempo transcurrido desde la menopausia (1-3 años; > 3 años) y sus valores iniciales de OMO lumbar (T > -1; y de -1 a -2.5). La administracion diaria de 2.5 mg de Bonviva® se tradujo en un aumento media de la OMO en un 3.1% en comparacion con placebo y del 1.9 % con los valores iniciales. En el grupo con placebo, al termino de dos años de estudio se aprecio una disminucion de aproximadamente 1 % de la OMO lumbar, confirmando que la disminucion de la masa ósea se acelera poco despues de la menopausia. lndependientemente del tiempo transcurrido desde la menopausia o del grado de OMO previo, el tratamiento con Bonviva® se tradujo en una respuesta de la OMO lumbar estadisticamente mayor que en los grupos con placebo en los cuatro estratos considerados. El 70% de las pacientes que recibieron Bonviva® respondieron al tratamiento (definiendo respuesta como un aumento de la OMO lumbar con respecto a los valores basales). Bonviva® tambien se asocio a un aumento media significativo de la OMO en la cadera de un 1.8% comparado con el grupo que recibio placebo y del 1.2 % en relacion a los valores basales. Tan solo un mes despues del comienzo del tratamiento se observo una reduccion clinicamente significativa en los marcadores bioquimicos de la resorcion ósea (CTX urinario). Propiedades farmacocineticas Los efectos farmacologicos del ibandronato no se relacionan directamente con las concentraciones sericas reales. Esto se demostro en varios estudios realizados en humanos y animales, en los que se demostro la eficacia equivalente del ibandronato despues de regimenes diarios o intermitentes, con un intervalo libre de farmaco de varias semanas (al menos 6 semanas en ratas, al menos 11 semanas en perros, al menos 30 dias en monos, y al menos 9.5 semanas en humanos) con una dosis total igual administrada durante este periodo. Absorcion Tras la administracion por vía oral, el acido ibandr6nico se absorbe rapidamente en el tracto gastrointestinal y sus concentraciones plasmaticas aumentan de forma proporcional a la dosis hasta los 50 mg, siendo su aumento mas que proporcional por encima de esta dosis. Las concentraciones plasmaticas maximas observadas alcanzan entre 0.5 y 2 horas (mediana = 1 hora) en ayuno y la biodisponibilidad absoluta fue del 0.6%. El grado de absorcion se ve deteriorado cuando se administra. La biodisponibilidad se reduce en casi 90% el ácido ibandronico es administrado junto. con un desayuno estandar en comparacion con la biodisponibilidad de los individuos en ayuno. No hay una reduccion significativa en la biodisponibilidad en tanto el acido ibandronico se ingiera 60 minutos antes de los alimentos. Tanto la biodisponibilidad coma el aumento de la OMO disminuyen cuando se administra un alimento solido o liquido en menos de 60 minutos despues de la dosis de Bonviva® Distribucion Tras la exposicion sistemica inicial, el acido ibandronico rapidamente se une a los huesos o se elimina con la orina. En los humanos, el volumen terminal aparente de distribucion es de por lo menos 90 L y la cantidad estimada de la dosis que alcanza los huesos es del 40 al 50% de la dosis en circulacion. La fijacion a las proteinas plasmaticas es escasa (aproximadamente 85% en concentraciones terapeuticas), por lo tanto, existe un bajo riesgo de interacciones farmacologicas debido al desplazamiento. Metabolismo No existe evidencia de que el acido ibandr6nico sea metabolizado en animales o en humanos. Eliminaci6n La fraccion sistemica disponible del acido ibandronico desaparece de la circulacion por absorcion ósea (40 - 50%) y el resto se elimina de forma inalterada en los riñones. La fraccion no absorbida del acido ibandronico se elimina de forma inalterada en las heces. El fndice aparentemente observado de las vidas medias es amplio y dependiente de la dosis y del ensayo de sensibilidad, pero la vida media terminal aparente se alcanza generalmente en el rango de 1 o a 72 horas. Las concentraciones plasmaticas iniciales descienden rapidamente, hasta llegar a un 10010 de los valores maximos en un plaza entre 3 y 8 horas despues de una administraci6n intravenosa u oral respectivamente. La depuracion total del acido ibandronico es baja, con valores promedio en el rango de 84 a 160 ml/min. La depuracion renal (cerca de 60 ml/min en mujeres posmenopausicas sanas) representa del 50 al 600/o de la depuraci6n total, y esta relacionada a la depuracion de la creatinina. Se considera que la diferencia entre la depuracion total aparente y la renal se debe al reflejo de la captacion ósea. Farmacocinetica en poblaciones especiales: Género. La biodisponibilidad y la farmacocinética del ácido ibandronico son similares tanto en hombres como en mujeres. No existe evidencia de diferencias interetnicas entre Asiaticos y Caucasicos que sean clinicamente relevantes en la disposici6n del acido ibandronico. Existen muy pocos datos disponibles de pacientes de origen africano. Pacientes con insuficiencia renal. La depuraci6n renal del acido ibandr6nico guarda una relacion lineal con la depuracion de creatinina (Cle,) en pacientes_ con diversos grados de insuficiencia renal. No es necesario ajustar la dosis en las pacientes con insuficiencia renal o hepatica leve o moderada (CL > 30 Ml/min.), como se mostro en el estudio BM 16549 donde la mayorfa de las pacientes cayo dentro de esta categoria. Las personas con insuficiencia renal grave (Cl < 30 ml/min) tratadas con 10 mg diarios de acido ibandronico por via oral durante 21 dias, presentaron concentraciones plasmaticas entre 2 y 3 veces mayores que las personas con funcion renal normal (Depuracion de creatinina = 129 ml/min). La depuracion total de creatinina se redujo a 44 ml/min en las personas con insuficiencia renal grave. Tras la administracion de 0.5 mg por via I.V., las cifras de depuracion disminuyeron un 67%, 77% y 50%, respectivamente en las personas con insuficiencia renal grave. Sin embargo, el consiguiente aumento de la exposicion no se asocio a una reduccion de la tolerabilidad. Pacientes con insuficiencia hepatica. No existen datos de farmacocinetica para el acido ibandronico en pacientes con dano hepatico. El higado no tiene un papel significativo en la depuracion del acido ibandronico debido a que no se metaboliza y a que es depurado por excrecion renal y absorbido en los huesos. Por esto no es necesario el ajuste de dosis en pacientes con dano hepatico. Adicionalmente, dado que la fijacion del acido ibandronico a las proteinas plasmaticas es baja en concentraciones terapeuticas (85%). la hipoproteinemia en enfermedades severas del higado es improbable para llevar a incrementos clinicamente significativos en la concentracion de plasma fibre. Ancianos En un analisis multivariable, se encontro que la edad no es un factor independiente de cualquiera de los parametros farmacocineticos estudiados. Como la funcion renal disminuye con la edad, este es el unico factor que debe tomarse en consideracion (ver Pacientes con insuficiencia renal).Niños: No exite datos del uso de Bonviva en pacientes menores de 18 años.
Contraindicaciones: Bonviva® esta contraindicado en pacientes con hipersensibilidad conocida al acido ibandronico o a cualquiera de las excipientes de la formula. Bonviva® esta contraindicado en pacientes con hipocalcemia no corregida. Como todos las bifosfonatos indicados en el tratamiento de la osteoporosis, la hipocalcemia preexistente necesita ser corregida antes de iniciar el tratamiento con Bonviva®. Como con varios bifosfonatos, Bonviva está contraindicado en pacientes con anomalias de esofago que retrasa el vencimiento. Bonviva está contraindicado en pacientes que no pueden permanecer de pie o sentados por mas de 60 minutos.
Precauciones generales: La hipocalcemia y otros disturbios óseos y del metabolismo mineral deben ser tratados efectivamente antes de iniciar la terapia con Bonviva. Las pacientes deben recibir un suplemento de calcio y de vitamina D si estos elementos no se consumen en la dieta. Los bifosfonatos administrados par via oral pueden causar irritacin local de la mucosa gastrointestinal superior. Debido a estos posibles efectos irritantes y al potencial de empeoramiento de la enfermedad subyacente, se debe tener precauci6n si Bonviva® se administra a pacientes con trastornos activos del aparato digestivo superior (por ejemplo, el conocido esofago de Barrett, disfagia, otras enfermedades esofagicas, gastritis, duodenitis o ulceras). Se han reportado eventos adversos, tales como esofagitis, ulceras esofagicas y erosiones esofagicas, en algunos casos graves y que requirieron hospitalizacion, raramente con sangrado o seguidas de estenosis esofagica o perforacion, en pacientes que recibian tratamiento con bifosfonatos orales. El riesgo de eventos adversos esofagicos graves parece ser mayor en pacientes que no cumplen con las instrucciones de dosificaci6n y/o que siguen tomando bifosfonatos par via oral despues de desarrollar sintomas que sugieran irritacion esofagica. Las pacientes deben prestar especial atencion y ser capaces de cumplir con las instrucciones de dosificacion. Los medicos deben estar atentos a cualquier signo o sintoma señalando una posible reaccion esofagica y las pacientes deben ser instruidos para interrumpir Bonviva® y buscar atencion medica si presentan disfagia, odinofagia, dolor retroesternal o aparicion o empeoramiento de la pirosis. Aunque no hay mayor riesgo, se observo en las ensayos clfnicos controlados que ha habido informes de ulceras gastricas y duodenales con el uso de bifosfonatos orales, algunas graves y con complicaciones despues de la comercializacion. Debido a que los AINE (antiinflamatorios no esteroideos) han sido asociados con la irritacion gastrointestinal, debe tomarse en consideracion cuando se administre una terapia oral concomitante con Bonviva®. Se ha reportado osteonecrosis de la mandibula en pacientes tratados con bifosfonatos. La mayoria de los casos han sido en pacientes con cancer bajo tratamientos odontologicos, pero algunos han ocurrido en pacientes con osteoporosis postmenopausica u otros diagnosticos. Los factores de riesgo conocidos para osteonecrosis de mandibula incluyen diagnostico de cancer, terapias concomitantes (por ej. quimioterapia, incluyendo inhibidores de la angiogenesis, radioterapia, corticosteroides) y des6rdenes comorbidos (por ej. anemia, coagulopatfa, infeccion, enfermedad dental pre-existente). Los casos han ocurrido en pacientes tratados con bifosfonatos administrados por via intravenosa pero algunos han sido en pacientes tratados por via oral. En los pacientes que desarrollan osteonecrosis de la mandibula mientras se encuentran en terapia con bifosfonato, la cirugia dental puede exacerbar esta condicion. En pacientes que requieren tratamientos odontologicos, no existen datos que sugieran que la interrupcion del tratamiento con bifosfonatos reduzca el riesgo de osteonecrosis mandibular. El juicio clinico del medico tratante debe guiar un plan de administracion para cada paciente, con base en la valoracion individual del riesgo-beneficio. No se han llevado a cabo estudios de los efectos de Bonviva® sabre la capacidad de conducir vehiculos y utilizar maquinaria. Tambien se han informado casos de osteonecrosis de otros sitios orofaciales, incluido el conducto auditivo externo, en pacientes tratados conbifosnatos incluyendo IBN. Los factores de riesgo son similares a los de la ONJ. Otros factores de riesgo pueden incluir trauma menor repetitivo (por ejemplo, uso habital de isopos de algodon). La posibilidad de osteonecrosis del conducto auditivo externo debe ser considerada en los pacientes que reciben bisfosfonatos que se presentan con síntomas del oido, incluyendo infecciones crónicas del oído.
Restricciones de uso durante el embarazo y la lactancia: Bon viva® no debe ser utilizado durante el em barazo y la lactancia. Embarazo: No se observaron evidencias de teratogenia ni toxicidad fetal directa del acido ibandronico en ratas y conejos tratados diariamente con acido ibandronico por vía oral, ni tampoco efectos adversos sobre el desarrollo de la descendencia (primera generaci6n, Fi) en ratas. Los efectos adversos del acido ibandronico en los estudios de toxicidad reproductiva con ratas fueron identicos a los observados con los bifosfonatos como grupo terapeutico. Entre ellos se cuentan la disminucion del numero de sitios de implante, la interferencia con el parto natural (distocia) y el aumento en las variaciones viscerales (sindrome ureteropielorrenal). Aun no se cuenta con estudios especificos para el regimen oral mensual. No existe experiencia clinica con Bonviva® en mujeres embarazadas. Lactancia: En ratas en periodo de lactancia tratadas con dosis de 0.08 mg/Kg/dia via IV de acido ibandronico, la mayor concentracion del farmaco medida en la leche fue de 8.1 ng/mL y se observo en las primeras 2 horas despues de la administracion de la infusion. Despues de 24 horas. la concentraci6n en la leche y el plasma fue similar, y correspondi6 al 5% de la concentracion medida despues de las primeras 2 horas de administracion. No se sabe si Bonviva es excretado en la leche humana.
Reacciones secundarias y adversas: Experiencia de estudios clfnicos Tratamiento de la osteoporosis posmenopausica dosis diaria. La seguridad de Bonviva® 2.5 mg, dosis diaria, fue evaluada en 1251 pacientes tratadas en 4 estudios clinicos placebo-controlados; 73% de estos pacientes participaron en el estudio de tratamiento pivote a 3 años (MF4411). El perfil de seguridad completo de Bonviva® 2.5 mg, dosis diaria, en estos estudios fue similar al del placebo. La proporci6n de pacientes que experimentaron alguna reaccion adversa al acido ibandr6nico. es decir, un evento adverso con una posible o probable relacion con el medicamento, en el estudio pivote de tratamiento. fue de 19.8% para Bonviva® 2.5 mg y 17.9% para el placebo. Dosis mensual: En un estudio a dos años, en mujeres postmenopausicas con osteoporosis (BMl 6549). se muestra que la seguridad de Bonviva® 150 mg una vez al mes y Bonviva® 2.5 mg diario es similar. La proporcion total de pacientes que experimentaron alguna reaccion adversa, ej: un evento adverso con una posible o probable relacion con el medicamento en estudio. fue de 22.7% y 25.0% para Bonviva® 150 mg una vez al mes y 21.5% y 22.5% para Bonviva® 2.5 mg. diariamente, despues de uno y dos años respectivamente. La mayorfa de estas reacciones adversas fueron de intensidad leve a moderada. La mayoria de las casos no condujeron a una suspension de la terapia. Las tablas 2 y 3 enlistan las reacciones adversas presentes en mas del 1% de las pacientes tratadas con Bonviva® 150 mg, una vez al mes o con 2.5 mg diarios en el estudio BMl 6549 y en pacientes tratadas con Bonviva® 2.5mg diarios en el estudio MF44 l 1. Las tablas muestran las reacciones adversas ocurridas en los dos estudios, que se presentaron en una mayor incidencia en comparacion con los pacientes tratados con placebo en el estudio MF 4411. Para cada grupo de frecuencias las efectos no deseados se presentan en orden descendiente de severidad. Los datos a un año de BMl 6549 se presentan en la Tabal 2 y las datos acumulados para los dos años de BM 16549 se presentan en la Tabla 3.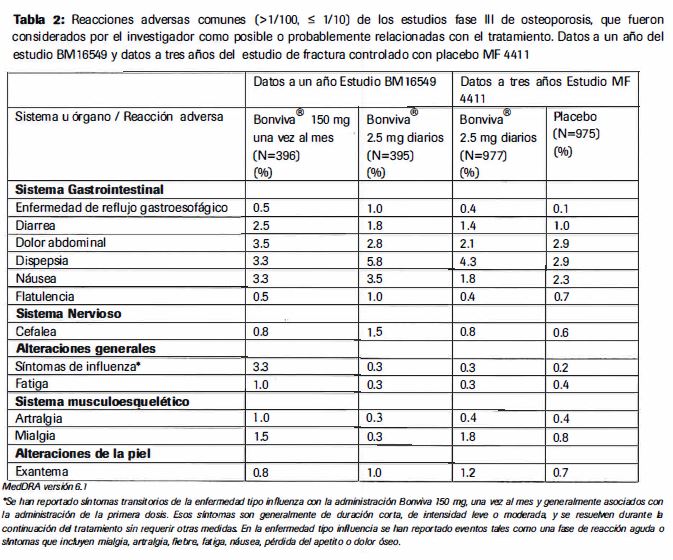
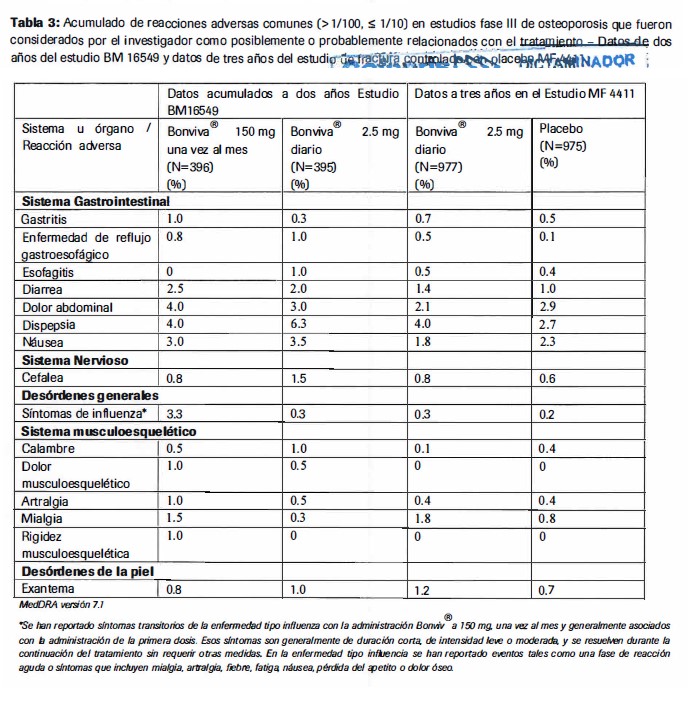
Reacciones adversas que se presentaron con una frecuencia igual o menos de 1% El siguiente listado proporciona informacion sobre reacciones adversas (consideradas por el investigador como posible o probablemente relacionadas con el tratamiento) reportadas en el estudio MF 4411 que ocurrieron mas frecuentemente con Bonviva® 2.5 mg diario que con placebo y el estudio BM 16549 que ocurrieron mas frecuentemente con Bonviva® 150 mg una vez al mes que con Bonviva® 2.5 mg diarios. Dentro de cada grupo de frecuencias, los efectos no deseados se presentan en orden descendente de severidad.
Dosis 1 vez al mes En el estudio del tratamiento una vez al mes, se incluyeron las pacientes con antecedentes de padecimientos gastrointestinales incluyendo pacientes con ulcera peptica sin sangrado u hospitalizacion reciente, y a las pacientes con dispepsia o reflujo controlado con medicamento. Para estas pacientes, no había diferencia en la incidencia de eventos adversos del tracto gastrointestinal superior con Bonviva® 150 mg una vez al mes comparado con el regimen diario de Bonviva® 2.5 mg. Prevencion de la osteoporosis posmenopausica: El perfil de seguridad de Bonviva® en el ensayo de prevenci6n de fase II/III MF 4499 (N=163 con 2.5 mg diarios de Bonviva®; N=159 con placebo) se comparo con los datos obtenidos en el estudio pivote de eficacia MF 4411 sin que aportara nuevos datos de seguridad. Post- comercializaci6n: Alteraciones musculoesqueleticas y tejido conectivo Se ha reportado caso de osteonecrosis de la mandibula y otros sitios oro-faciales, incluyendo el conducto auditivo externo, en pacientes tratados con acido lbandronico muy raramente. Alteraciones oculares Se han reportado eventos de inflamacion ocular coma uveitis, epiescleritis y escleritis con el uso de bifosfonatos, incluyendo acido ibandronico. En algunos casos, estos eventos no se resolvieron, aun con el retiro del bifosfonato. Alteraciones del sistema lnmune Se han reportado casos de reacciones choque anafilacticos, incluyendo eventos fatales en pacientes tratados con acido lbandronico. Se han reportado reacciones alergicas incluyendo exacerbaci6n asmatica. Se han reportado reacciones adversas cutaneas graves que incluyen el Sindrome de StevensJohnson, eritema multiforme y dermatitis ampollosa. Lesiones, envenenamiento y complicaciones del procedimiento Se han informado fracturas diafisiarias y subtrocantericas atipicas femorales con terapia de bifosfonatos, incluyendo ibandronato; sin embargo, no se ha establecido una causalidad.
Interacciones medicamentosas y de otro género: Interacciones con los alimentos Los productos que contienen calcio y otros cationes polivalentes (tales como aluminio, magnesio, hierro), incluyendo la leche y otros alimentos, pueden interferir con la absorcion de Bonviva® segun se ha observado en los estudios con animales. Por lo tanto, la ingestion de tales productos o alimentos, se debe separar al menos 60 minutos de la ultima administraci6n oral. lnteracciones farmacologicas Es probable que los suplementos de calcio, los antiacidos y algunos medicamentos orales que contienen cationes polivalentes (tales como aluminio, magnesia, hierro) puedan interferir con la absorcion de Bonviva®. Por lo tanto, las pacientes deben esperar 60 minutos desde la ultima administracion oral Bonviva® antes de tomar otros medicamentos orales. Los estudios de interacciones farmacocineticas en mujeres posmenopausicas han demostrado la inexistencia de interaccion potencial con tamoxifeno o con la terapia de reemplazo hormonal (estrogenos). Tampoco se han observado interacciones con la administracion simultanea de melfalano/prednisolona en pacientes con mieloma multiple. En voluntarios sanos hombres y en mujeres posmenopausicas, la ranitidina administrada intravenosamente causo un incremento en la biodisponibilidad del acido ibandronico de alrededor del 20%, probablemente como resultado de la reduccion de la acidez gastrica. Sin embargo, ya que el incremento esta dentro del rango normal de la biodisponibilidad del acido ibandronico, no se requiere ajuste de dosis cuando Bonviva® es administrado con antagonistas de H2 o cualquier otro farmaco que incrementa el pH gastrico. En relacion a la disposicion, no se considera probable ninguna interaccion de trascendencia clinica, puesto que el acido ibandronico no inhibe las principales isoenzimas hepaticas humanas del sistema P450 y tampoco induce dicho sistema hepatico en las ratas. Ademas, la fijacion de las proteinas plasmaticas es baja en concentraciones terapeuticas de modo que parece poco probable que el acido ibandronico pueda desplazar a otros farmacos. El acido ibandronico es eliminado por excrecion renal solamente y no sufre de ninguna biotransformaci6n. Segun parece, la vía de secrecion no incluye los sistemas conocidos de transporte basicos o acidos implicados en la excrecion de otros farmacos. En un estudio clfnico de 1 año de duracion realizado en mujeres posmenopausIcas con osteoporosis (BMl 6549), la incidencia de eventos del tracto gastrointestinal superior a quienes se administro concomitantemente aspirina o AINE fue similar a la de las pacientes que tomaron Bonviva® 2.5 mg diario o 150 mg una vez al mes. De las 1500 pacientes que participaron en el estudio clinico BMl 6549 en el que se compararon las dosis mensual y diaria de acido ibandronico, el 14% de las pacientes usaron bloqueadores de histamina (HJ o inhibidores de la bomba de protones. Entre estas pacientes, la incidencia de eventos del tracto gastrointestinal superior en los pacientes tratados con Bonviva 150 mg, una vez al mes, fue similar a las pacientes tratadas con Bonviva 2.5 mg diariamente.
Alteraciones en los resultados de pruebas de laboratorio: En un estudio pivote a 3 anos con 2.5mg diarios de Bonviva® (MF4411) no se apreciaron diferencias en comparaci6n con el placebo en cuanto a las alteraciones analfticas indicativas de disfunci6n hepatica o renal, alteraci6n del sistema hematico, hipocalcemia o hipofosfatemia. De igual manera, no se encontraron diferencias entre grupos despues de uno y dos anos en el estudio BM 16549
Precauciones y relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Los efectos toxicos que se observaron en animales fueron unicamente con exposiciones consideradas suficientemente superiors a la exposicion humana maxima siendo por lo tanto indicativas de ascasas tracendencias para el uso clínico. No se ha observado ningun indicio de riesgo carcinogénetico ni genotóxico.
Dosis y vía de administración: La dosis recomendada de Bonviva® para el tratamiento y la prevencion de la osteoporosis es de una tableta de 150 mg una vez al mes. La tableta de 150 mg debe tomarse preferentemente en la misma fecha cada mes. Bonviva® debe administrarse al menos 60 minutos antes del primer alimento o bebida del dia (con excepcion del agua) o cualquier otro medicamento oral (incluyendo los suplementos de calcio): Las tabletas deben deglutirse enteros acompanadas de un vaso lleno de agua simple (180 a 240 m L) mientras la paciente esta en una posicion vertical, sentada o de pie. Las pacientes no deben acostarse hasta 60 minutos despues de haber ingerido Bonviva® El agua simple es la unica bebida con la que se debe tomar Bonviva® Adviertase que ciertas bebidas con gas pueden tener concentraciones elevadas de calcio y por lo tanto no deben ser utilizadas. Las pacientes no deben masticar ni chupar las tabletas debido a un alto riesgo de que se produzcan ulceras orofaringeas. Las pacientes deben recibir suplemento de calcio o vitamina D si el consumo de estas sustancias en la dieta fuera insuficiente. En caso de olvidar una dosis mensual, las pacientes deben tomar un tableta de Bonviva® de 150 mg en la manana siguiente al recordarlo excepto si la siguiente dosis debe tomarse en un plazo no mayor a 7 dias. Las pacientes deben volver a tomar la dosis mensual en la fecha originalmente designada. Si la proxima dosis esta programada dentro de un plazo de 7 dias, las pacientes deben esperar hasta su proxima dosis y continuar tomando una tableta al mes como el esquema original. Las pacientes no deben tomar dos tabletas de 150 mg en la misma semana. Pautas posologicas especiales. Pacientes con insuficiencia hepatica: No es necesario hacer ajuste de la dosis. Pacientes con insuficiencia renal: No es necesario hacer ajuste de la dosis para las pacientes con insuficiencia renal leve o moderada con cifras de CL > , L 30 mL/min. En el caso de Cl < 30 mL/min, la decision de administrar Bonviva® se debe basar en una valoraci6n individual del indice riesgo/beneficio. Ancianos: No se considera necesario ajuste de la dosificacion. Niños: La seguridad y la eficacia no se han establecido en pacientes menores de 18 años de edad.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: Hasta la fecha, no se tienen casos concretos de sobredosificacion con Bonviva®. Sin embargo, la sobredosis oral puede ocasionar una exacerbacion de reacciones adversas gastrointestinales, como el malestar estomacal, ardor, esofagitis, gastritis o ulceras. Puede administrarse leche o antiacidos para fijar Bonviva® Dado el riesgo de irritacion esofagica, no debe provocarse el vomito y se aconseja mantener a las pacientes en posicion completamente vertical.
Presentaciones: Caja con envase de burbuja con 1 tableta de 150 mg.
Recomendaciones sobre almacenamiento: Conservase a no mas de 30°c. lnstrucciones especiales para su uso, manejo y desecho. Desecho de medicamento caduco o no usado. La liberacion de productos farmaceuticos en el medio ambiente debe ser minimizada. Los medicamentos no se deben tirar por los desagues y la eliminacion a traves de la basura debe ser evitada.
Leyendas de protección: Su venta requiere receta medica Este medicamento debera ser prescrito por medicos especialistas No se deje al alcance de los niños No se use durante el embarazo ni la lactancia Literatura exclusiva para medicos Reporte las sospechas de reacciones adversas al correo farmacovigilancia@cofepris.gob.mx. La seguridad del paciente es una responsabilidad compartida, Roche recibe sus reportes de sospechas de reacciones adversas a traves del Centro de Informacion Médica y la Unidad Farmacovigilancia. Contáctanos: mexico.info@roche.com
Nombre y domicilio del laboratorio: ROCHE
Número de registro del medicamento: 020M2005 SSA IV
BONVIVA®
MOKSHA8
Solución inyectable
Denominación genérica: Acido ibandrónico.
Forma farmacéutica y formulación: Solución inyectable. Cada jeringa precargada contiene: ibandronato monosódico monohidratado equivalente a 3 mg de ácido ibandrónico. Vehículo cbp 3 ml.
Indicaciones terapéuticas: BONVIVA® inyectable está indicado en el tratamiento de la osteoporosis postmenopáusica, para reducir el riesgo de fracturas.
Farmacocinética y farmacodinamia: Propiedades farmacocinéticas: los efectos farmacológicos del ibandronato no están relacionados directamente con las concentraciones en el plasma, ya que el sitio de acción es el tejido óseo. Esto fue demostrado en varios estudios en animales y en humanos, en los cuales la eficacia equivalente del ibandronato fue confirmada tanto después de la administración diaria como de manera intermitente al mantener intervalos sin medicamento de varias semanas (al menos 6 semanas en ratas, 11 semanas en perros, 30 días en monos y al menos 9,5 semanas en humanos), administrando la misma dosis total durante este período. Las concentraciones de ibandronato en plasma se incrementaron de una manera proporcional a la dosis, después de la administración intravenosa de 0,5 mg a 6 mg. Distribución: después de la administración, el ibandronato se adhiere rápidamente al hueso o es excretado en la orina. En humanos, el volumen aparente de distribución terminal es de al menos 90 l y la cantidad de dosis que se alcanza en el hueso se estima de 40-50% de la dosis circulante. La adherencia a proteínas plasmáticas humanas es baja (aproximadamente 85% unida a concentraciones terapéuticas); por lo tanto hay un bajo potencial de interacción medicamento-medicamento debido al desplazamiento. Metabolismo: no hay evidencia de que el ibandro