BRILINTA®
ASTRAZENECA
Denominación genérica: Ticagrelor.
Forma farmacéutica y formulación: Tabletas. Cada tableta contiene: Ticagrelor 90 mg. Excipiente cbp una tableta.
Indicaciones terapéuticas: BRILINTA® está indicado para la prevención de eventos trombóticos (muerte cardiovascular, infarto de miocardio y accidente cerebrovascular) en pacientes con Síndrome Coronario Agudo ([SCA] angina inestable, infarto de miocardio sin elevación del segmento ST [NSTEMI] o infarto de miocardio con elevación del segmento ST [STEMI]) incluyendo pacientes tratados médicamente o aquellos tratados con intervención coronaria percutánea (PCI) o puentes de derivación aorto-coronarios (CABG).
Farmacocinética y farmacodinamia: Propiedades farmacocinéticas: Generales: Ticagrelor presenta una farmacocinética lineal y la exposición a BRILINTA® y al metabolito activo (AR-C124910XX) son aproximadamente proporcionales a la dosis. Absorción: La absorción de ticagrelor es rápida con una mediana de tmáx de aproximadamente 1.5 horas. La formación del principal metabolito circulante AR-C124910XX (también activo) es rápida con una mediana de tmáx de aproximadamente 2.5 horas. Los valores de Cmáx (concentración máxima) y ABC (área bajo la curva) de Ticagrelor y del metabolito activo aumentaron en forma aproximadamente proporcional con la dosis en el rango estudiado (30-1260 mg). La biodisponibilidad media absoluta de ticagrelor se estimó en un 36%, (rango 25.4% a 64.0%). La ingesta de una comida rica en grasas no tuvo efecto sobre los valores de Cmáx de ticagrelor o de ABC del metabolito activo, pero produjeron un aumento del 21% en el ABC de ticagrelor y una disminución del 22% en la Cmáx del metabolito activo. Estos pequeños cambios se consideran de significancia clínica mínima; por lo tanto, BRILINTA® puede administrarse con las comidas o alejado de ellas. El ticagrelor como tabletas trituradas mezcladas con agua, administradas oralmente o a través de una sonda nasogástrica en el estómago, es bioequivalente a las tabletas enteras (ABC y Cmax. de 80-125% para el ticagrelor y el metabolito activo). La exposición inicial (0.5 y 1 hora despés de la dosis) de las tabletas trituradas de ticagrelor mezcladas con agua fue mayor en comparación con las tabletas enteras, con un perfil de concentración generalmente idéntico a partir de entonces (de 2 a 48 horas). Distribución: El volumen de distribución en estado estacionario de ticagrelor es 87.5 L. Ticagrelor y el metabolito activo se unen ampliamente a las proteínas plasmáticas humanas ( > 99.0%). Metabolismo: La CYP3A es la principal enzima responsable del metabolismo de ticagrelor y de la formación del metabolito activo y sus interacciones con otros sustratos de la CYP3A varían entre la activación y la inhibición. Ticagrelor y el metabolito activo son inhibidores débiles de la glicoproteína P. El principal metabolito de ticagrelor es AR-C124910XX, que también fue activo cuando se evaluó por la unión in vitro al receptor de ADP plaquetario P2Y12. La exposición sistémica al metabolito activo es de aproximadamente el 30-40% de la obtenida para ticagrelor. Excreción: La vía primaria de eliminación de ticagrelor es el metabolismo hepático. Cuando se administra ticagrelor radiomarcado, la media de recuperación de la radioactividad es de aproximadamente el 84% (57.8% en las heces, 26.5% en la orina). La recuperación de ticagrelor y del metabolito activo en la orina fueron ambas menores del 1% de la dosis. La vía primaria de eliminación del metabolito activo es mayormente la excreción biliar. La media de t1/2 fue de aproximadamente 6.9 horas (rango 4.5-12.8 horas) para ticagrelor y 8.6 horas (rango 6.5-12.8 horas) para el metabolito activo. Poblaciones especiales: Pacientes de edad avanzada: Se observó mayor exposición a Ticagrelor [aproximadamente 60% tanto para Cmáx como para ABC (área bajo la curva)] y al metabolito activo (aproximadamente 50% tanto para Cmáx como para ABC) en sujetos de edad avanzada (≥ 65 años) comparados con sujetos más jóvenes. Esta diferencia no se considera clínicamente significativa. (véase Dosis y vía de administración). Pacientes pediátricos: BRILINTA® no se ha evaluado en la población pediátrica. (véase Dosis y vía de administración). Sexo: Se observó una mayor exposición a Ticagrelor [aproximadamente 52 y 37% para Cmáx (distribución) y ABC (área bajo la curva), respectivamente] y al metabolito activo (aproximadamente 50% tanto para Cmáx como para ABC) en mujeres comparada con hombres. Estas diferencias no se consideran clínicamente significativas. Insuficiencia renal: La exposición a ticagrelor fue aproximadamente 20% más baja y la exposición al metabolito activo fue aproximadamente 17% en pacientes en pacientes con función renal normal. El efecto IPA (Inhibición de la Agregación Plaquetaria) de ticagrelor fue similar entre los dos grupos, sin embargo se observó más variabilidad en la respuesta individual de pacientes con insuficiencia renal severa. No es necesario el ajuste de la dosis en pacientes con insuficiencia renal. No se dispone de información respecto del tratamiento de los pacientes en diálisis renal. (véase Dosis y vía de administración). Insuficiencia hepática: Los valores de Cmáx y ABC de ticagrelor fueron 12% y 23% más altos en pacientes con insuficiencia hepática leve comparados con sujetos sanos respectivamente, sin embargo el efecto IPA de ticagrelor fue similar entre los dos grupos. No se necesita ajuste de la dosis en pacientes con insuficiencia hepática leve. Ticagrelor no ha sido estudiado en pacientes con insuficiencia hepática moderada o severa. (véase Dosis y vía de administración). Raza: Los pacientes de origen asiático tienen una biodisponibilidad media 39% más alta en comparación con los pacientes caucásicos. Los pacientes de raza negra tuvieron una biodisponibilidad 18% más baja de ticagrelor en comparación con los pacientes caucásicos. En estudios de farmacología clínica, la exposición (Cmáx y ABC) a ticagrelor en sujetos japoneses fue aproximadamente 40% (20% luego del ajuste por peso corporal) más alta que en los caucásicos. Propiedades farmacodinámicas: Mecanismo de acción: BRILINTA® contiene ticagrelor, un miembro de la clase química de las ciclopentiltriazolpirimidinas (CPTP), que es un antagonista del receptor P2Y12 oral de acción directa, de unión reversible y selectiva, que impide la activación y la agregación de las plaquetas dependientes del P2Y12 mediado por la adenosina difosfato (ADP). El ticagrelor no impide la unión de ADP, pero cuando se enlaza al receptor P2Y12 evita la transducción de la señal inducida por la ADP. Dado que las plaquetas participan en la iniciación y la evolución de las complicaciones trombóticas de la enfermedad ateroeclerótica, se demostró que la inhibición de la función plaquetaria reduce el riesgo de eventos cardiovasculares como muerte, infarto de miocardio o accidente cerebrovascular. El ticagrelor tiene un mecanismo de acción adicional que aumenta los niveles locales de adenosina endógena inhibiendo el transportador de equilibrio de nucleósidos 1 (ENT-1). La adenosina se forma localmente en los sitios donde existe hipoxia y daño tisular mediante la degradación de la adenosina trifosfato (ATP) y difosfato (ADP) liberada. Dado que la degradación de la adenosina está esencialmente restringida al espacio intracelular, la inhibición de ENT-1 mediante el ticagrelor prolonga la vida media de la adenosina y, por ende, aumenta su concentración extracelular local y ofrece una respuesta mejorada de la adenosina local. El ticagrelor no tiene efecto directo clínicamente significativo sobre los receptores de adenosina (A1, A2A, A2B, A3) y no se metaboliza en adenosina. Se ha documentado que la adenosina tiene una serie de efectos que incluyen: vasodilatación, cardioprotección, inhibición plaquetaria, modulación de la inflamación e inducción de disnea, lo cual puede contribuir al perfil clínico del ticagrelor. Efectos farmacodinámicos: Comienzo de la acción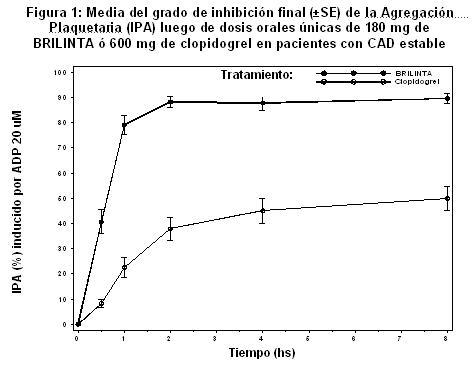
En pacientes con enfermedad arterial coronaria estable tratados con ASA, BRILINTA® demuestra un comienzo rápido del efecto farmacológico a través de la media de la Inhibición de la Agregación Plaquetaria (IPA) a las 0.5 horas después de una dosis de carga de 180 mg de alrededor de 41%, con el máximo de efecto de IPA de 87.9% a 89.6% a las 2-4 horas post dosis. Noventa por ciento de los pacientes tuvieron un grado final de IPA > 70% a las 2 horas post dosis. El efecto alto de IPA de BRILINTA® de entre 87%-89% se mantuvo entre 2-8 horas.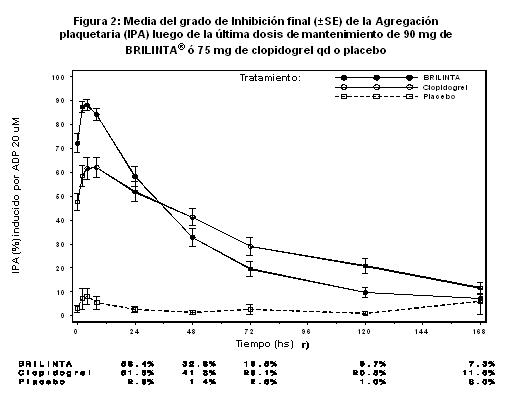
Finalización del efecto: Luego de que las concentraciones de BRILINTA® y del metabolito activo declinan a un nivel menor al requerido para la saturación del receptor, la IPA disminuye gradualmente con la disminución de las concentraciones plasmáticas. Dado que BRILINTA® se une reversiblemente, la recuperación de la función plaquetaria no depende del reemplazo de plaquetas. BRILINTA® tiene un índice de finalización de la IPA más rápido que clopidogrel, determinado por la declinación de la finalización desde las 4-72 horas posteriores a la última dosis (véase Precauciones generales). La mediana del grado final de IPA medido luego de la última dosis de BRILINTA® es aproximadamente 20-30% mayor con BRILINTA® que con clopidogrel. Sin embargo, a las 24 horas posdosis, el % de IPA es similar entre BRILINTA® y clopidogrel, y es más bajo para BRILINTA® a partir de las 72 horas hasta los 7 días en comparación a clopidogrel. La media de % de IPA con BRILINTA® a las 72 horas (Día 3) posteriores a la última dosis fue comparable a la de clopidogrel en el Día 5, y el % de IPA con BRILINTA® en el Día 5 fue similar al de clopidogrel en el Día 7, lo cual no es diferente estadísticamente del placebo. Respuesta a BRILINTA®: La IPA inducida por BRILINTA® tiene menos variabilidad en las concentraciones plasmáticas máximas de BRILINTA® y del metabolito activo observado con la dosis de 90 mg bd en comparación con la de clopidogrel. Los pacientes con enfermedad arterial coronaria estable predeterminados para tener una baja respuesta IPA a clopidogrel (no-respuesta), y que reciben dosis concomitantes de ASA, mostraron una respuesta IPA media mayor luego de la administración de BRILINTA® en comparación a clopidogrel. En los de no-respuesta a clopidogrel, se observó una respuesta IPA más alta y consistente con BRILINTA®. El tratamiento con BRILINTA® resultó en una IPA consistentemente más alta en comparación con clopidogrel, y esto fue evidente posterior a la administración, tanto en los que responden como en los que no responden. Resultados de cambio de medicamento: El cambio de clopidogrel a BRILINTA® resulta en un incremento absoluto de IPA del 26.4% y el cambio de BRILINTA® a clopidogrel resulta en una disminución absoluta de IPA del 24.5%. Los pacientes pueden cambiar de clopidogrel a BRILINTA® sin interrupción del efecto antiplaquetario. Mecanismo de la adenosina (ENT-1): El ticagrelor aumentó las concentraciones plasmáticas de la adenosina en pacientes con síndrome coronario agudo (SCA) y se ha demostrado que aumenta varias respuestas fisiológicas a la adenosina. La adenosina es un vasodilatador; se ha demostrado que el ticagrelor aumenta el flujo sanguíneo coronario inducido por la adenosina en voluntarios sanos y pacientes con SCA. La adenosina es un inhibidor plaquetario endógeno; se ha demostrado que el ticagrelor aumenta la inhibición de la agregación plaquetaria mediada por la adenosina junto con la inhibición plaquetaria debido a su antagonismo de P2Y12. La adenosina se ha vinculado con el efecto cardioprotector del preacondicionamiento; se ha demostrado que el ticagrelor reduce el tamaño del infarto a través de un mecanismo mediado por la adenosina en un modelo de rata de lesión de revascularización. La adenosina también induce disnea; se ha demostrado que el ticagrelor aumenta la disnea inducida por la adenosina en voluntarios sanos. Por lo tanto, esposible que la disnea observada en algunos pacientes que usan ticagrelor sea mediada total o parcialmente por la adenosina. Eficacia clínica: La evidencia clínica de la eficacia de BRILINTA® se deriva del estudio PLATO [PLATelet Inhibition and Patient Outcomes], una comparación de BRILINTA® con clopidogrel, ambos administrados en combinación con ácido acetil salicílico (ASA) y otros tratamientos estándares. El estudio PLATO fue un estudio de 18,624 pacientes aleatorizados, doble ciego, de grupos paralelos, de fase III, de eficacia y seguridad de BRILINTA® comparado con clopidogrel para la prevención de eventos vasculares en pacientes con Síndromes Coronarios Agudos (SICA) (angina inestable, Infarto de miocardio sin elevación del ST [NSTEMI] o Infarto de miocardio con elevación del ST [STEMI]. El estudio incluyó pacientes que se presentaban dentro de las 24 horas del inicio del episodio más reciente de dolor torácico o síntomas. Los pacientes fueron aleatorizados para recibir clopidogrel (75 mg una vez por día, con una dosis de carga inicial de 300 mg si no se había administrado tratamiento previo con una tienopiridina. Se permitía una dosis de carga adicional de 300 mg a discreción del investigador), o una dosis de carga de 180 mg de BRILINTA® seguida de una dosis de mantenimiento de 90 mg de BRILINTA® dos veces por día. Los pacientes podían haber sido tratados médicamente, haber sido tratados con intervención coronaria percutánea (PCI) o puentes de derivación aorto-coronarios (CABG).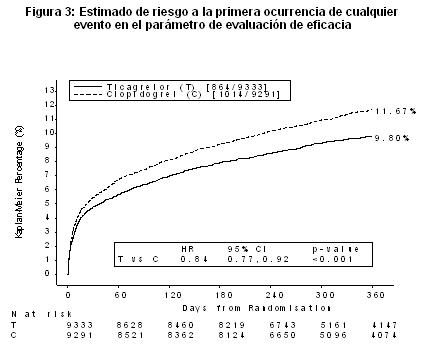
BRILINTA® redujo la aparición del parámetro de evaluación compuesta primaria en comparación con clopidogrel tanto en la población UA/NSTEMI como en la STEMI.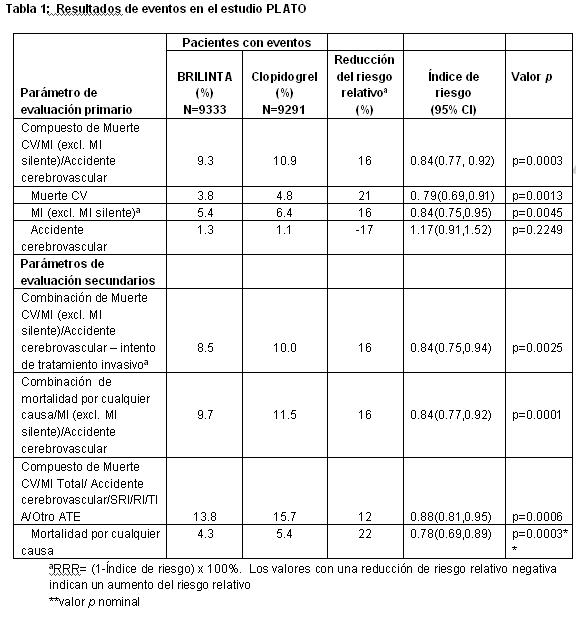
BRILINTA® es superior a clopidogrel en la prevención de eventos trombóticos (RRR 16%, ARR 1,9%, NNT =54) del parámetro de evaluación compuesto de eficacia (muerte cardiovascular (CV), infarto de miocardio (MI) o accidente cerebrovascular) durante 12 meses. La diferencia entre los tratamientos fue explicada por la muerte cardiovascular y el infarto de miocardio sin diferencias en los accidentes cerebrovasculares. BRILINTA® demostró una reducción estadísticamente significativa del riesgo relativo del 16% (ARR 1.1%) para MI y una reducción del riesgo relativo del 21% (ARR 1.1%) para muerte CV. El tratamiento de 91 pacientes con BRILINTA® en lugar de clopidogrel prevendrá 1 muerte CV. BRILINTA® mostró superioridad frente a clopidogrel en la prevención del objetivo primario (muerte cardiovascular [CV], infarto de miocardio [MI o un accidente cerebrovascular). Este resultado apareció tempranamente (reducción del riesgo absoluto [RRA] 0.6% y reducción del riesgo relativo [RRR] de 12% a los 30 días), con un efecto constante en el tratamiento durante un periodo completo de 12 meses, produciendo un RRA 1.9% por año con un RRR del 16 %. Esto sugiere que es apropiado para el tratamiento durante al menos 12 meses (véase Dosis y vía de administración). En el estudio PLATO se llevaron a cabo un gran número de comparaciones de subgrupos para el parámetro de evaluación primario de eficacia, con el fin de evaluar la robustez y consistencia del beneficio global. El efecto del tratamiento de BRILINTA® sobre clopidogrel parece ser consistente entre los múltiples subgrupos de tratamiento por características demográficas incluyendo peso, sexo, antecedentes médicos, terapia concomitante, y por diagnóstico final del evento [STEMI, NSTEMI, y angina inestable (UA)]. Se observó una débil interacción significativa del tratamiento regionalmente, en que la HR para el parámetro de evaluación primario favorece a BRILINTA® en el resto del mundo pero favorece a clopidogrel en América del Norte, lo cual representa aproximadamente el 10% de la población global estudiada (valor de p para la interacción=0.045). Esta aparente interacción regional del tratamiento observada en PLATO posiblemente podría atribuirse a la casualidad, al menos en parte. Análisis adicionales sugieren que la eficacia de clopidogrel en relación con BRILINTA® se asocia con dosis de ASA durante la terapia de mantenimiento. Los datos muestran una mayor eficacia de ticagrelor en comparación con clopridogrel cuando se utiliza con una dosis baja de mantenimiento de ASA (75-150 mg). La eficacia relativa de ticagrelor frente a clopidogrel cuando se utiliza con altas dosis de ASA ( > 300 mg) es más incierta. Con base en esta relación observada entre la dosis de mantenimiento de ASA y la eficacia relativa de ticagrelor en comparación con clopidogrel, se recomienda que BRILINTA® se utilice con una dosis de mantenimiento baja de ASA 75-150 mg (véase Precauciones generales y Dosis y vía de administración). Los beneficios asociados con BRILINTA® también fueron independientes del uso de otras terapias cardiovasculares agudas y a largo plazo, incluyendo heparina, heparina de bajo peso molecular (LMWH), inhibidores GpIIb/IIIa intravenosos, fármacos hipolipemiantes, beta bloqueantes, inhibidores de la enzima convertidora de angiotensina (IECA), antagonistas del receptor de la angiotensina II (ARAII), e inhibidores de la bomba de protones (IBP) (véase Interacciones medicamentosas y de otro género). BRILINTA® demostró una reducción de riesgo relativo (RRR) estadísticamente significativo en el parámetro de evaluación compuesto de muerte cardiovascular (CV), infarto de miocardio (MI) y accidente cerebrovascular en pacientes con SCA programados para someterse a tratamiento invasivo (RRR 16%, ARR 1.7%, p=0.0025). En un análisis exploratorio, BRILINTA® demostró una reducción del riesgo relativo del parámetro de evaluación primario compuesto en pacientes con SCA programados para someterse a tratamiento médico (RRR 15%, ARR 2.3%, p nominal=0.0444). En forma consistente con el parámetro de evaluación primario del estudio, el efecto en estos dos grupos fue justificado por muerte CV y MI, sin efecto sobre el accidente cerebrovascular. En los pacientes a los que se les coloca stent, hubo numéricamente menos trombosis definidas por stent, entre pacientes tratados con ticagrelor en comparación con los tratados con clopidogrel (73 vs. 107, RRR 32%, ARR 0.6%, p nominal =0.0123). BRILINTA® demostró una RRR estadísticamente significativa del 16% (ARR 2.1%) para el compuesto de mortalidad de cualquier causa, MI y accidente cerebrovascular comparado con clopidogrel. Se evaluó el parámetro de evaluación secundario (mortalidad de cualquier causa). BRILINTA® demostró una RRR del 22% para mortalidad por cualquier causa en comparación con clopidogrel a un nivel de significancia nominal de p=0.0003 y una ARR del 1.4%. Subestudio Holter: Para estudiar la aparición de pausas ventriculares y otros episodios de arritmias durante el estudio PLATO, los investigadores realizaron un monitoreo con Holter en un subgrupo de casi 3000 pacientes, de los cuales aproximadamente 2000 tuvieron registros tanto en la fase aguda de su SCA como después de un mes. La variable primaria de interés fue la aparición de pausas ventriculares ≥3 segundos. Más pacientes tuvieron pausas ventriculares con BRILINTA®(6.0%) que con clopidogrel (3.5%) en la fase aguda; y 2.2% y 1.6% respectivamente luego de 1 mes. Más pacientes tuvieron pausas ventriculares con BRILINTA® que con clopidogrel, sin embargo, no hubo consecuencias clínicas adversas asociadas con esta diferencia (incluyendo colocación de marcapasos) en esta población de pacientes. Ticagrelor es activo por vía oral. A diferencia de clopidogrel, no requiere la actividad de la enzima CYP450 para inhibir la agregación plaquetaria. Los polimorfismos en el gen que codifica la enzima CYP450, 2C19 puede afectar la eficacia de clopidogrel. El polimorfismo en el gen que codifica para el transporte de P-glicoproteína (ABCB1) puede afectar a la eficacia tanto de clopidogrel y ticagrelor. En el estudio PLATO, 10,285 pacientes proporcionaron muestras genéticas para la determinación del genotipo CPY2C19 y ABCB1 loci. Su análisis proporcionó estas asociaciones de agrupaciones de genotipo, sobre la eficacia y seguridad de los resultados del estudio PLATO. La superioridad de BRILINTA® sobre clopidogrel no se ve afectada significativamente en los pacientes con el genotipo CYP2C19. BRILINTA® reduce los eventos CV mayores en comparación con clopidogrel de forma independiente del genotipo CYP2C19. La tasa de eventos para BRILINTA® no varió con el genotipo CYP2C19. En el grupo tratado con clopidogrel, los portadores del alelo CYP2C19 Pérdida de Función (LOF) aumentaron las tasas de eventos primarios extremos en comparación con los no portadores. Al igual que en el estudio global PLATO, el sangrado mayor total no fue diferente entre BRILINTA® y clopidogrel, independientemente del genotipo CYP2C19, aunque los pacientes con uno o más alelos Ganadores de Función (GOF) en clopidogrel tuvieron la tasa mayor de sangrado. Al igual que en el estudio global PLATO, el sangrado no-CABG se incrementó con BRILINTA® en comparación con clopidogrel en pacientes con un alelo CYP2C19 LOF. El sangrado no-CABG fue similar con BRILINTA® y clopidogrel en pacientes sin alelo CYP2C19 LOF. Análisis combinado de eficacia y seguridad: Un análisis combinado de eficacia y seguridad (muerte CV, MI, accidente cerebrovascular, o sangrado "Mayor Total" según PLATO) avala el beneficio clínico de ticagrelor comparado con clopidogrel (RRR 8%, ARR 1.4%, HR 0.92; p=0.0257) durante 12 meses luego de los eventos de SCA.
Contraindicaciones: BRILINTA® no debe administrarse a pacientes con antecedentes de hipersensibilidad a ticagrelor o a alguno de los excipientes, ni tampoco en casos de sangrado patológico activo, historia de hemorragia intracraneal o insuficiencia hepática grave (véase Reaciones secundarias y adversas.
Precauciones generales: Riesgo de sangrado: Como con otros agentes antiplaquetarios, el uso de BRILINTA® en pacientes con riesgo conocido de aumento de sangrado, se debe ponderar contra el beneficio, en términos de prevención de eventos trombóticos. Si está clínicamente indicado, BRILINTA® debe utilizarse con precaución en los siguientes grupos de pacientes: Se debe considerar lo siguiente: Pacientes propensos al sangrado (por ej. debido a traumatismo reciente, cirugía reciente, sangrado gastrointestinal reciente o activo, o insuficiencia hepática moderada). El uso de BRILINTA® está contraindicado en pacientes con sangrado patológico activo y en aquéllos con antecedentes de hemorragia intracraneal e insuficiencia hepática grave (véase Contraindicaciones). Pacientes con administración concomitante de productos medicinales que puedan aumentar el riesgo de sangrado (por ej., medicamentos anti-inflamatorios no esteroideos (AINEs), anticoagulantes orales y/o fibrinolíticos dentro de las 24 horas de la administración de BRILINTA®). No existen datos con BRILINTA® con respecto al beneficio hemostático de las transfusiones plaquetarias; el ticagrelor circulante puede inhibir las plaquetas transfundidas. Dado que la coadministración de BRILINTA® con desmopresina no disminuyó el patrón del tiempo de sangrado, es improbable que la desmopresina sea efectiva en el manejo de los eventos clínicos de sangrado. La terapia antifibrinolítica (ácido aminocaproico o ácido tranexámico) y/o el factor VIIa recombinante pueden aumentar la hemostasia. BRILINTA® puede reanudarse luego de haber identificado y controlado la causa del sangrado. Cirugía: Si un paciente requiere cirugía, los médicos deben considerar el perfil clínico de cada paciente así como los beneficios y riesgos de continuar con la terapia antiplaquetaria al determinar cuando debe realizarse la interrupción del tratamiento con BRILINTA®. Dada la unión reversible de BRILINTA®, la restauración de la agregación plaquetaria se produce más rápido con BRILINTA® en comparación con clopidogrel. En el estudio OFFSET, la media de la Inhibición de la Agregación Plaquetaria (IPA) con BRILINTA®a las 72 horas postdosis fue comparable a la media de IPA con clopidogrel a las 120 horas post-dosis. Una recuperación de la acción más rápida puede predecir un riesgo reducido de complicaciones de sangrado, por ej, en situaciones en las que la terapia antiplaquetaria debe interrumpirse temporalmente debido a cirugía o traumatismo. En los pacientes del estudio PLATO sometidos a CABG, BRILINTA® tuvo un índice similar de sangrados mayores en comparación con clopidogrel, todos los días después de suspender la terapia excepto en el Día 1 en que BRILINTA® tuvo un índice más alto de mayor sangrado. Si un paciente se va a someter a cirugía programada y no se desea conservar el efecto antiplaquetario, BRILINTA® debe descontinuarse 5 días antes de la cirugía. (véase Propiedades farmacodinámicas). Pacientes con insuficiencia hepática moderada: Se recomienda precaución en pacientes con insuficiencia hepática moderada ya que BRILINTA® no se ha estudiado en estos pacientes. El uso de BRILINTA® está contraindicado en pacientes con insuficiencia hepática grave (véase Contraindicaciones). Pacientes con riesgo de eventos bradicárdicos: Debido a observaciones de pausas ventriculares principalmente asintomáticas en un estudio clínico temprano, los pacientes con riesgo mayor de eventos bradicárdicos (por ej. pacientes sin marcapasos que tienen síndrome de seno enfermo, bloqueo AV de segundo o de tercer grado o síncope por bradicardia) fueron excluidos del estudio principal que evaluaba la seguridad y eficacia de BRILINTA®. Por lo tanto, debido a la experiencia clínica limitada en estos pacientes, se aconseja tener precaución (véase Propiedades farmacodinámicas). Disnea: Se reporta disnea en los pacientes tratados con BRILINTA® (aproximadamente 13.8%), generalmente la intensidad es de leve a moderada y con frecuencia se resuelve sin necesidad de interrumpir el tratamiento, (véase Reacciones secundarias y adversas). El mecanismo aún no ha sido esclarecido. Si un paciente reporta que presenta disnea reciente, prolongada o empeoramiento, se debe investigar a fondo y si no se tolera el tratamiento con BRILINTA® se debe interrumpir. Se informó de Disnea en 13.3% de pacientes tratados con BRILINTA® y en 7.8% de pacientes tratados con clopidogrel. Los investigadores consideraron que solo en el 2.2 % de pacientes la disnea tuvo relación causal con el tratamiento de BRILINTA®. Habitualmente ésta, fue de intensidad leve a moderada y habitualmente se resolvió sin la necesidad de interrumpir el tratamiento. Los pacientes con Asma o Enfermedad Pulmonar Obstructiva Crónica pueden tener un incremento en el riesgo absoluto para presentar disnea con BRILINTA®. Por tanto, ticagrelor debería ser utilizado con precaución en pacientes con historia de Asma o Enfermedad Pulmonar Obstructiva Crónica. El mecanismo aun no se ha esclarecido. Si un paciente informa de nuevos episodios de disnea o que ésta se prolongue o empeore, se deberán investigar otras posibles causas y si el tratamiento con BRILINTA® no es tolerado, éste deberá ser interrumpido. Otros: Basados en la relación observada en PLATO entre la dosis de mantenimiento de ASA y la eficacia relativa de ticagrelor en comparación con clopidogrel, la administración concomitante de ticagrelor y dosis altas de mantenimiento de ASA ( > 300 mg) no se recomiendan (véase Propiedades farmacodinámicas). La administración concomitante de BRILINTA® con inhibidores potentes de la CYP3A4 (por ej, ketoconazol, claritromicina, nefazadona, ritonavir, y atanazavir) debe evitarse, ya que la misma puede producir un aumento sustancial en la exposición a BRILINTA® (véase Interacciones medicamentosas y de otro género). Interrupción del tratamiento: Los pacientes que requieran interrumpir BRILINTA® tienen un aumento de riesgo de eventos cardiacos. Debe evitarse la interrupción prematura del tratamiento. Si debe interrumpirse BRILINTA® temporalmente debido a un evento adverso, su uso debe reanudarse tan pronto como sea posible cuando los beneficios superen a los riesgos del evento adverso o cuando el evento adverso se haya resuelto (véase Dosis y vía de administración). Efectos en la habilidad para conducir u operar maquinaria: No se han realizado estudios sobre los efectos de BRILINTA® sobre la capacidad de conducir y utilizar maquinaria. Durante el tratamiento de los Síndromes Coronarios Agudos, se han reportado mareos y confusión. Por lo tanto, los pacientes que experimenten estos síntomas deben tener precaución al conducir o utilizar maquinarias.
Restricciones de uso durante el embarazo y la lactancia: No se han llevado a cabo estudios clínicos en mujeres embarazadas o en mujeres durante la lactancia. Se dispone de datos clínicos limitados sobre la exposición a BRILINTA® durante el embarazo. Los estudios en animales no indican efectos perjudiciales directos respecto del embarazo, del desarrollo embrionario/fetal, del parto ni del desarrollo postnatal. Ticagrelor no tuvo efecto sobre la fertilidad masculina ni femenina (véase Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre fertilidad). Dado que los estudios de reproducción en animales no siempre son predictivos de la respuesta en seres humanos, ticagrelor debe utilizarse durante el embarazo solamente cuando el beneficio potencial para la madre justifique algún riesgo potencial para el feto. Lactancia: No se conoce si este medicamento se excreta en humanos en la leche materna. Los estudios en ratas mostraron que ticagrelor y sus metabolitos activos se excretan en la leche. No se recomienda el uso de BRILINTA® durante la lactancia.
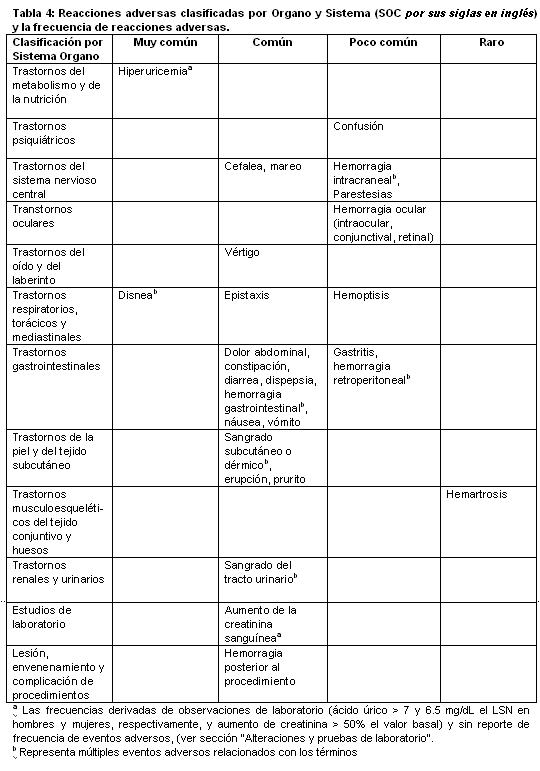
Reacciones secundarias y adversas: Resumen del perfil de seguridad: El perfil de seguridad de BRILINTA® ha sido evaluado en los resultados de dos grandes estudios fase 3 (PLATO y PEGASUS) que incluyeron a más de 39.000 pacientes. Las reacciones adversas relevantes, observadas en estos estudios se discuten a continuación. La seguridad de BRILINTA® en pacientes con síndromes coronarios agudos (UA, NSTEMI y STEMI) fue evaluada en el estudio PLATO, el cual comparó pacientes tratados con BRILINTA® 90 mg dos veces al día con pacientes tratados con clopidogrel 75 mg una vez al día, ambos administrados en combinación con ASA y otras terapias estándar. La mediana de duración del tratamiento con BRILINTA® fue de 277 días. Durante el período de tratamiento, los pacientes del estudio PLATO con BRILINTA® tuvieron una mayor incidencia de interrupción debida a los eventos adversos que en clopidogrel (7.4% vs. 5.4%). La seguridad de BRILINTA® en pacientes con historia de IM (el IM ocurrido al menos hace un año) y riesgo alto de desarrollar un evento trombótico, fue evaluada en el estudio PEGASUS, el cual comparó a pacientes tratados con BRILINTA® 60 mg dos veces al día o 90 mg dos veces al día combinado con ASA, con la terapia solo con ASA y otras terapias estándar. Las reacciones adversas reportadas más comunmente en pacientes tratados con ticagrelor fueron sangrado y disnea. Lista tabulada en reacciones adversas: Las reacciones adversas de los estudios clínicos PLATO y PEGASUS con BRILINTA® (Tabla 1) están listadas por la Clasificación de Órganos y Sistemas (SOC por sus siglas en inglés) del MedDRA y por categoría de frecuencia. Dentro de cada SOC y categoría de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad. Las categorías de frecuencia están definidas de acuerdo a las siguientes convenciones: Muy común (≥ 1/10), Común (≥ 1/100 a < 1/10), Poco común (≥ 1/1000 a < 1/100), Rara (≥ 1/10000 a < 1/1000), Muy rara ( < 1/10000), No conocida (no se puede estimar con los datos disponibles).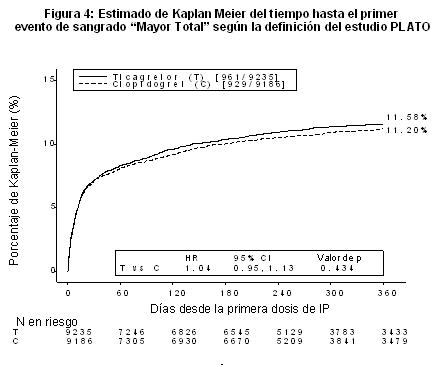
Descripción de reacciones adversas seleccionadas: Sangrado: Se utilizaron las siguientes definiciones de sangrado en el estudio PLATO: Mayor Fatal/Amenaza la vida: Sangrado fatal o intracraneal o intrapericárdico con taponado cardíaco, o con choque hipovolémico o hipotensión severa debida al sangrado y que requiere presores o cirugía o clínicamente aparente o sangrado aparente asociado con una reducción de la hemoglobina de más de > 50 g/l, o transfución de 4 o más unidades (sangre completa o PRBCs) para el sangrado. Otro Mayor: Significativamente incapacitante (p. ej., intraocular con pérdida permanente de la visión), o clínicamente aparente o sangrado aparente asociado con una reducción en la hemoglobina de 30 a 50 g/l, o transfusión de 2-3 unidades (sangre completa o PRBCs) para el sangrado. Menor: Requiere intervención médica para detener o tratar el sangrado (p. ej., epistaxis que requiere visita a la instalación médica para taponar). Sangrados mínimos incluyeron todos los sangrados; estos datos se recolectaron pero no se adjudicaron. Los eventos de sangrado reportados en el PLATO fueron también mapeados a la escala TIMI (Trombolisis en Infarto al Miocardio), para facilitar la comparación con otros estudios similares. TIMI Mayor está definido como un sangrado clinicamente evidente asociado con una caída en la hemoglobina > 50g/l, o hemorragia intracraneal, y el TIMI Menor está definido como un sangrado evidente asociado con una caída en la hemoglobina de 30 g/l pero ≤ 50 g/l. El resultado global de los eventos de sangrado en el estudio PLATO se muestra en la Figura 4 y la Tabla 3.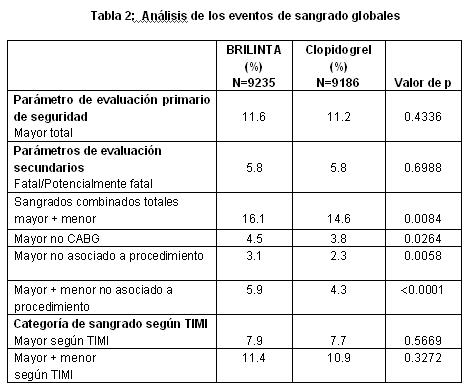
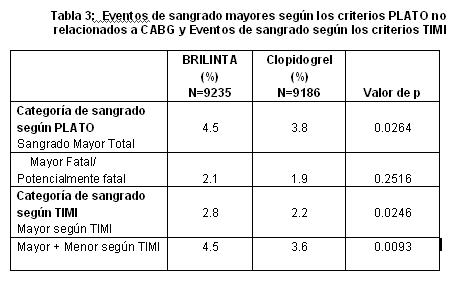
Sangrado no relacionado con algún procedimiento: Según se muestra en la Tabla 2 en "Mayor" y "Mayor + Menor" definidos por PLATO, el sangrado no debido al procedimiento fue más frecuente con BRILINTA®. La interrupción del tratamiento debida al sangrado no debido al procedimiento fue más común para BRILINTA® (2.9%) que para el clopidogrel (1.2%, p≤0.001). Las ubicaciones clínicamente importantes para el sangrado "Mayor + Menor" en el orden clasificado por frecuencia fueron (BRILINTA® vs clopidogrel): Intracraneal (27 vs 14 eventos), pericárdica (11 vs 11), retroperitoneal (3 vs 3), intraocular (2 vs 4) e intrarticular (2 vs 1). Otras ubicaciones comunes en el orden clasificado de frecuencias fueron: gastrointestinal (170 vs 135 eventos), epistaxis (115 vs 61), urinario (45 vs 37), subcutáneo/dérmico (43 vs 38) y hemóptisis (13 vs 7). No hubo ninguna diferencia con BRILINTA® en comparación con el clopidogrel para sangrado fatal no debido al procedimiento. El sangrado gastrointestinal "Mayor Fatal/Amenaza para la vida" fue el mismo con BRILINTA® y clopidogrel, con más eventos fatales numéricamente para el clopidogrel (5) que para el BRILINTA® (ninguno). Numéricamente, hubo má eventos de sangrado "Mayor Fatal/Amenaza para la vida" intracraneales no debidos al procedimiento con BRILINATA® (n=27 eventos en 26 pacientes, 0.3%) que con el clopidogrel (n=14 eventos, 0.2%), de los cuales, 11 eventos de sangrado con BRILINTA® y 1 con clopidogrel fueron fatales. Las características en la línea basal incluyeron edad, género, peso, raza, región geográfica, historia médica, condiciones concurrentes y terapia concomitante fueron evaluadas para explorar algún incremento en el riesgo de sangrado con BRILINTA®. No se identificó ningún grupo de riesgo particular para algún subconjunto de sangrado. Disnea: El el PLATO, se reportaron eventos adversos de disnea en 13.8% de los pacientes que tomaban ticagrelor 90 mg dos veces al día y en 7.8% en pacientes que tomaban clopidogrel 75 mg una vez al día. La mayoría de los eventos adversos de disnea reportados fueron de intensidad leve a moderada y por lo general se resolvieron sin necesidad de la interrupción del tratamiento. La disnea fue habitualmente reportada en la fase inicial del tratamiento y 87% de los pacientes que reportaron disnea experimentaron un solo episodio. Eventos adversos graves de disnea fueron reportados en un 0.7% de los pacientes que tomaban ticagrelor y en 0.4% de los que tomaban clopidogrel. Los pacientes que reportaron disnea tendieron a ser más ancianos y con más frecuencia tuvieron disnea, CHP, EPOC o asma en la línea basal. Los datos del estudio PLATO no sugieren que la mayor frecuencia con el BRILINTA® se deba a una nueva enfermedad de corazón o de pulmón o al agravamiento de estas. No hubo ninguna indicación de un efecto adverso del BRILINTA® sobre la función pulmonar.
Interacciones medicamentosas y de otro género: Interacciones farmacológicas: Efectos de otros fármacos sobre BRILINTA®: Medicamentos metabolizados por CYP3A4: Ketoconazol (Inhibidores potentes de CYP3A4): La administración concomitante de ketoconazol con ticagrelor aumentó los valores de Cmáx y de ABC de ticagrelor a 2.4 y 7.3 veces respectivamente. Los valores de Cmáx y ABC del metabolito activo se redujeron un 89% y un 56% respectivamente. Se esperaría que otros inhibidores potentes de CYP3A4 (claritromicina, nefazadona, ritonavir y atanazavir) tuvieran efectos similares y no deberían administrarse concomitantemente con BRILINTA® (véase Precauciones generales). Diltiazem (Inhibidores moderados de CYP3A4): La administración concomitante de ticagrelor y diltiazem aumentó los valores de Cmáx de ticagrelor en un 69% y los de ABC en un 174% y disminuyó los valores de Cmáx del metabolito activo en un 38% y el ABC del metabolito activo no se modificó. No hubo efecto de ticagrelor sobre los niveles plasmáticos de diltiazem. Otros inhibidores moderados de CYP3A4 (por ej., amprenavir, aprepitant, eritromicina, fluconazol y verapamil) también pueden administrarse concomitantemente con BRILINTA®. Rifampicina y otros inductores de CYP3A4: La administración concomitante de rifampicina con ticagrelor disminuyó los valores de Cmáx y ABC de ticagrelor en un 73% y un 86%, respectivamente. La Cmáx del metabolito activo no se modificó y el ABC disminuyó en un 46%, respectivamente. Se esperaría que otros inductores de CYP3A4 (por ej. dexametasona, fenitoína, carbamacepina y fenobarbital) disminuyan también la exposición a ticagrelor y pueden reducir la eficacia de BRILINTA®. Ciclosporina (PgP e inhibidor CYP3A): La coadministración de ciclosporina (600 mg) con ticagrelor, incrementa la Cmax y el ABC en 2.3 y 2.