COMBIGAN®-D
ALLERGAN
Denominación genérica: Tartrato de Brimonidina / Maleato de Timolol.
Forma farmacéutica y formulación: Solución. Cada ml contiene: Tartrato de brimonidina 2mg. Maleato de timolol 6.8 mg* equivalentes a 5 mg de Timolol. Vehículo c.b.p. 1ml. * La concentración actual es 6.8 mg/mL de maleato de timolol el cual corresponde a 0.5% timolol base libre.
Indicaciones terapéuticas: COMBIGAN®-D está indicado para la reducción de la presión intraocular (PIO), en pacientes con glaucoma de ángulo abierto o hipertensión ocular que requieren tratamiento complementario o de reemplazo debido a un control inadecuado de la PIO o que responden de forma insuficiente a los beta bloqueadores tópicos. Farmacocinética y farmacodinamia: Propiedades farmacodinamicas: Mecanismo de acción: COMBIGAN®-D contiene dos sustancias activas: tartrato de brimonidina y maleato de timolol. Estos dos componentes disminuyen la PIO elevada mediante mecanismos de acción complementarios y el efecto combinado da como resultado una mayor reducción de la PIO comparado con los compuestos administrados por separado. COMBIGAN®-D tiene un rápido inicio de acción. El tartrato de brimonidina es un agonista potente del receptor alfa-2 adrenérgico que es 1000 veces más selectivo para el receptor alfa-2 adrenérgico. La afinidad al receptor adrenérgico humano 1-alfa y 2-alfa es de ~200 nM y ~ 2nM, respectivamente. Esta selectividad da como resultado la ausencia de midriasis y la ausencia de vasoconstricción de los micro vasos. La administración tópica del tartrato de brimonidina disminuye la PIO en humanos. Cuando se utiliza como está indicado, el tartrato de brimonidina tiene la acción de reducir la PIO elevada con mínimo efecto en los parámetros cardiovasculares. Los estudios fluorométricos en animales y humanos sugieren que el tartrato de brimonidina tiene un mecanismo de acción dual. La brimonidina reduce la PIO mediante la supresión de la producción del humor acuoso y mejora el drenado uveoscleral. El timolol es un agente bloqueador del receptor adrenérgico beta1 y beta2 (no selectivo), que no tiene actividad simpaticomimética intrínseca significativa, depresión directa en el miocardio o anestesia local (estabilizadora de membrana). El Timolol reduce la PIO mediante la reducción de la formación del humor acuoso. Tanto la brimonidina como el timolol tienen un rápido inicio de acción, con un efecto máximo de hipotensión ocular visto a las 2 horas después de administrar brimonidina y de una a dos horas para timolol. La reducción significante de la presión intraocular puede ser mantenida por periodos de hasta 12 horas para brimonidina y de 24 horas para una dosis simple de timolol. Estudios clínicos: La PIO elevada representa un factor de riesgo significativo en la perdida glaucomatosa del campo visual. Entre mayor sea el nivel de la PIO, mayor será la posibilidad de daño al nervio óptico y la pérdida del campo visual. Cinco estudios clínicos de E.U.A y dos estudios Europeos fueron realizados para evaluar la eficacia y seguridad de COMBIGAN®-D y demostraron perfiles de seguridad aceptables. Un estudio clínico fase 2 de 7 días (N=73) fue realizado para evaluar la seguridad, eficacia y tolerabilidad de COMBIGAN®-D administrado 2 veces al día, comparado con tartrato de brimonidina al 0.2% administrado 3 veces al día y timolol al 0.5% administrado dos veces al día, cada uno administrado por 7 días. El estudio demostró que la administración a corto plazo de COMBIGAN®-D fue bien tolerado con un perfil de seguridad similar al timolol y a la brimonidina y propició la reducción estadísticamente significativa y clínicamente relevante de la PIO de hasta 7.8mm Hg respecto al valor basal en pacientes con glaucoma o hipertensión ocular. Dos estudios clínicos de 3 meses (con extensión a un año) (N=1,159) fueron realizados para determinar la eficacia y la seguridad de COMBIGAN®-D administrado dos veces al día comparado con brimonidina administrado 3 veces al día y timolol administrado dos veces al día en pacientes con glaucoma o hipertensión ocular. Los análisis agrupados de 12 meses de dos estudios fundamentales indican que COMBIGAN®-D administrado dos veces al día controló la PIO diurna durante el estudio y fue superior, para timolol al 0.5% y brimonidina al 0.2% 3 veces al día, en reducir la PIO elevada en pacientes con glaucoma o hipertensión ocular. La media disminuyó con COMBIGAN®-D desde el valor basal de la PIO de 4.4 a 7.6 mm Hg, durante 12 meses de seguimiento, de 2.7 a 5.5 con brimonidina, y de 3.9 a 6.2 mm Hg con timolol. Las reducciones medias de la PIO fueron significativamente mayores con COMBIGAN®-D comparado con timolol a todas las mediciones (P≤0.002) y con brimonidina a las 8 am, 10 am y 3 pm (P < 0.001) pero no a las 5 pm. La incidencia de los eventos adversos relacionados al tratamiento en el grupo de combinación fija de COMBIGAN®-D fue menor que en el grupo de brimonidina (P=0.006) pero mayor que en grupo de timolol (P < 0.001). En general, en relación al tratamiento, la incidencia de los eventos adversos relacionados a la alergia conjuntival e inflamación, los cuales están típicamente asociados con brimonidina e incluye cualquier combinación de eventos tales como hiperemia conjuntival, prurito ocular, y conjuntivitis folicular, fue significativamente menor en el grupo de COMBIGAN®-D que en el grupo de brimonidina (26.0% vs 39.8%; P < 0.001). El perfil de seguridad general de la terapia fija de brimonidina-timolol en este estudio fue consistente con los resultados de los estudios previos de brimonidina y timolol como monoterapias. No se presentaron problemas de seguridad con la terapia fija de brimonidina-timolol que no se hayan observado con los principios activos por separado. Un estudio clínico de 4 semanas (N=432) fue realizado para determinar la seguridad y eficacia de COMBIGAN®-D administrado 2 veces al día comparado con brimonidina administrado 3 veces al día simultáneamente con timolol administrado 2 veces al día y ALPHAGAN® al 0.2% administrado 3 veces al día en pacientes sin tratamiento previo con glaucoma e hipertensión ocular. El tratamiento con COMBIGAN®-D durante 28 días fue bien tolerado y redujo efectivamente la PIO en pacientes sin tratamiento previo con glaucoma o hipertensión ocular, exponiendo a los pacientes a menores eventos adversos en comparación con el tratamiento concomitante de brimonidina y timolol, con aproximadamente un 33% de reducción en exposición a la brimonidina. El efecto reductor de la PIO de COMBIGAN®-D no fue inferior al tratamiento concomitante en el 2o y 3er puntos de tiempo del seguimiento, hora 0 (~8 am,, punto de tiempo durante el efecto del fármaco) y hora 2 (~10 am, punto de tiempo del efecto máximo del fármaco). El único punto de tiempo en donde la reducción de la PIO con COMBIGAN®-D fue menor (aproximadamente 1 mm Hg) que el visto con la administración concomitante de timolol al 0.5% dos veces al día y brimonidina al 0.2% 3 veces al día fue en la hora 8, efecto máximo de la dosis adicional de medio día del componente Alphagan® TID del tratamiento concomitante. En el día 28, la media de la PIO diurna promedio en todos los puntos de tiempo (Horas 0, 2 y 8) fue de 17.1, 16.8 y 18.8 mm Hg en los grupos de la combinación, concomitante, y de Alphagan®, respectivamente. La media diurna PIO no fue estadística y significativamente diferente entre el grupo de la combinación y el grupo concomitante. El grupo Alphagan® TID fue usado como brazo de validación y se comporto como se esperaba. Los dos estudios europeos fase 3 enfocados en pacientes en quienes la PIO mostró estar controlada inadecuadamente bajo mono terapia. Los estudios que comparan la terapia de COMBIGAN®-D con las terapias de brimonidina dos veces al día o timolol dos veces al día y con la terapia concomitante de brimonidina y timolol dos veces al día. Ambos estudios tuvieron una duración de 12 semanas. En el estudio 190342-506T (N=589), a las 12 semanas, la disminución media del valor basal de la PIO fue significativamente mayor con COMBIGAN®-D que con brimonidina en las horas 0 (efecto mínimo) y 2 (efecto máximo) y fue significativamente mayor con COMBIGAN®-D que con timolol en el punto máximo (hora 2) (-4.3 a -5.0 mm Hg en el grupo de COMBIGAN®-D, -2.7 a -4.0 en el grupo de brimonidina y -3.0 a -3.9 mm Hg en el grupo de timolol). Los eventos adversos en general y los eventos adversos relacionados al tratamiento fueron mayores en el grupo de COMBIGAN®-D que en el grupo de timolol. No hubo diferencia en las comparaciones por pares para la incidencia de cualquier evento adverso relacionado al tratamiento. A las 12 semanas en el estudio 190342-507T (N=371), la media cambió desde el valor basal de la PIO de -4.9 a -5.3 mm Hg en ambos grupos de tratamiento. El grupo de COMBIGAN®-D no fue inferior a la terapia concomitante en todas las visitas de seguimiento. Los eventos adversos relacionados al tratamiento no fueron estadística y significativamente diferentes entre los dos grupos. Propiedades Farmacocinéticas: Farmacocinética clínica: En un estudio cruzado de tres periodos, se determinaron las concentraciones plasmáticas de la brimonidina y timolol en 16 sujetos sanos a los que se les administró dos veces al día durante 7 días (combinación, brimonidina, y timolol dos veces al día). No hubieron diferencias estadísticamente significativas en la AUC (área bajo la curva) de la brimonidina y timolol entre COMBIGAN®-D y los respectivos tratamientos de monoterapia. Los valores de C max (concentración máxima) promedio de brimonidina para COMBIGAN®-D [0.0327 ± 0.015(media ± DS, N=15)] y para la solución oftálmica de tartrato de brimonidina al 0.2% [0.0347 ± 0.0226 ng/mL (N=16)] no indicaron una diferencia aparente. Los valores de C max promedio de timolol para COMBIGAN ®-D fueron 0.406 ± 0.216 (media ± D.S., n = 15) y 0.507 ± 0.269 ng/ml (n = 14) para timolol al 0.5%. A pesar de que la Cmax de timolol fue aproximadamente 20% menor en el tratamiento con COMBIGAN®-D, la diferencia no fue estadísticamente significativa (p = 0.088). Después de la aplicación ocular de COMBIGAN®-D en sujetos humanos sanos, la vida media sistémica aparente fue de siete horas. El monitoreo terapéutico del fármaco fue realizado en dos estudios fase 3 en un subgrupo de pacientes. Las concentraciones plasmáticas de brimonidina y timolol después de administrar COMBIGAN®-D dos veces al día fueron 30-40% menores que sus respectivos valores en monoterapia. En el caso de la brimonidina, la diferencia parece ser debida a la dosificación de dos veces al día para COMBIGAN®-D y la dosificación de tres veces al día para ALPHAGAN®. Las bajas concentraciones plasmáticas de timolol observadas con la aplicación de COMBIGAN®-D, comparado con timolol al 0.5%, son resultado de la lenta absorción sistémica del timolol, debido aparentemente a la baja concentración de cloruro de benzalconio en COMBIGAN®-D y no a una interacción medicamentosa entre la brimonidina y el timolol. Después de una administración oral en animales y humanos, la brimonidina y sus metabolitos fueron rápidamente eliminados de la circulación sistémica mediante la excreción vía urinaria. Una pequeña dosis fue excretada como fármaco original en la orina. Aproximadamente el 87% de una dosis radioactiva administrada vía oral fue eliminada en humanos dentro de 120 horas, encontrado el 74% en la orina. Después de una dosificación tópica en humanos, la brimonidina se une aproximadamente en un 29% a las proteínas plasmáticas. La proporción de la radioactividad total en sangre-plasma fue de aproximadamente de 1 después de una dosificación oral de 14C-brimonidina. La brimonidina fue extensamente metabolizada en humanos sistémicamente. Se metaboliza principalmente por el hígado, más probablemente por el citocromo P450 y por la aldehído-oxidasa. Las principales vías metabólicas de la brimonidina son la oxidación del carbono alfa de la mitad de la quinoxalina al derivado quinoxalin-2,3-dione y la escisión oxidativa del anillo de imidazolina al arilguanidina. Los metabolitos de quinoxalol son metabolizados por glucoronidación. La rápida eliminación por el metabolismo sistémico de metabolitos polares parece limitar la distribución en el tejido y la exposición del cuerpo a la brimonidina. Después de una administración ocular de solución de brimonidina al 0.2% dos veces al día por 10 días, las concentraciones plasmáticas fueron menores (Cmax media fue de 0.06 ng/mL). El área bajo la curva del tiempo de concentración plasmática después de 12 horas en estado estacionario (AUC0-12h) fue de 0.31 ng*hr/mL, comparado con 0.23 ng*hr/mL después de la primera dosis. La vida media aparente en la circulación sistémica fue de aproximadamente 3 horas en humanos después de una dosificación tópica. La exposición sistémica de timolol después de una administración oral en hombres ha sido bien caracterizada. El timolol administrado oralmente se absorbe rápido y casi por completo (~90% disponibilidad). Las concentraciones plasmáticas de timolol detectables se presentan dentro de la media hora después de la administración y las concentraciones plasmáticas máximas se presentan dentro de la primera a segunda hora después de la administración. La vida media de eliminación aparente del timolol en el plasma es de 4 horas. La vida media permanece esencialmente sin cambios en pacientes con insuficiencia renal moderada. El timolol es parcialmente metabolizado por el hígado y el timolol y sus metabolitos son excretados por el riñón. El timolol no se une extensamente a las proteínas plasmáticas (~60%). Después de una administración oral, el timolol está sujeto al metabolismo de primer paso moderado (~50%). Sólo una pequeña cantidad del fármaco aparece en la orina sin cambios, junto con sus metabolitos después de la administración oral.
Contraindicaciones: COMBIGAN®-D está contraindicado en: Pacientes con enfermedad reactiva de las vías respiratorias incluyendo: asma bronquial, historial de asma bronquial o enfermedad pulmonar obstructiva crónica severa. Pacientes con bradicardia sinusal, enfermedad del nódulo sinusal, bloqueo del nódulo sinoauricular, bloqueo aurículo ventricular de segundo y tercer grado no controlado con marcapasos; insuficiencia cardiaca evidente, choque cardiogénico. Pacientes con hipersensibilidad a cualquiera de los componentes de este medicamento. Pacientes que estén recibiendo terapia inhibidora de mono amino oxidasa (MAO). No se use en menores de 18 años.
Precauciones generales: Como cualquier otro tipo de medicamento oftálmico tópico, los principios activos (tartrato de brimonidina y timolol) de COMBIGAN®-D pueden ser absorbidos sistémicamente. No se ha observado un incremento en la absorción sistémica individual de los principios activos. COMBIGAN®-D no ha sido estudiado en menores de 18 años. Sin embargo, en un estudio de tres meses fase 3 de niños entre 2-7 años con glaucoma inadecuadamente controlado por beta bloqueadores, el uso de tartrato de brimonidina en solución oftálmica al 0.2% provocó una alta incidencia y severidad de somnolencia en niños de 2 años en adelante, especialmente en aquellos con un peso ≤20kg. Función renal y hepática: No se ha estudiado COMBIGAN®-D en pacientes con insuficiencia renal o hepática; estos pacientes se deben tratar con precaución. Enfermedades cardiacas: COMBIGAN®-D debe ser usado con precaución en pacientes con enfermedad cardiovascular (ejemplo., enfermedad cardiaca coronaria, angina de Prinzmetal e insuficiencia cardiaca) e hipotensión. En los pacientes que tienen un historial de enfermedades cardiovasculares se deben monitorear los signos de evolución de estas enfermedades. Debido a su efecto negativo en el tiempo de conducción, los beta-bloqueadores deben ser administrados con precaución a pacientes con bloqueo cardiaco de primer grado. Padecimientos vasculares: Los pacientes con alteración/enfermedad severa de la circulación periferica (ejemplo: fenómeno de Raynaud) deben ser tratados con precaución. Enfermedad obstructiva pulmonar: Los pacientes con enfermedad pulmonar obstructiva crónica (ejemplo: bronquitis crónica, enfisema) de severidad leve o moderada no deben, en general, recibir productos que contengan beta bloqueadores, incluyendo COMBIGAN®-D; sin embargo, si COMBIGAN®-D se considera necesario para estos pacientes, debe ser administrado con precaución. Anafilaxia: Mientras se administran beta bloqueadores, los pacientes con historial de atopia o historial de reacciones anafilácticas severas a una variedad de alérgenos, pueden ser más reactivos a exposiciones repetidas con tales alergenos. Dichos pacientes pueden no responder a dosis usuales de epinefrina usadas para tratar reacciones anafilácticas. Diabetes mellitus: Los agentes bloqueadores beta-adrenérgicos deben ser administrados con precaución en pacientes con hipoglucemia espontánea o en pacientes diabéticos (especialmente aquellos con diabetes lábil). Debido a que los agentes bloqueadores de los receptores beta-adrenérgicos pueden enmascarar los signos y síntomas de hipoglucemia aguda. Hipertiroidismo: los agentes bloqueadores beta-adrenérgicos pueden enmascarar signos de hipertiroidismo. Alteraciones corneales: Los beta-bloqueadores oftálmicos pueden provocar sequedad ocular. Los pacientes con alteraciones corneales deben ser tratados con precaución. Desprendimiento coroideo: Se ha reportado desprendimiento coroideo después de procedimientos de filtración con la administración de terapia supresora acuosa (ejemplo: timolol). Otros agentes beta-bloqueadores: Se debe tener precaución al usar concomitantemente con agentes bloqueadores sistémicos beta-adrenérgicos debido al efecto aditivo potencial en los beta-bloqueadores sistémicos. Se debe monitorear de cerca la respuesta de estos pacientes. No se recomienda el uso de dos agentes bloqueadores tópicos beta-adrenérgicos. Anestesia quirúrgica: Los beta-bloqueadores oftálmicos pueden afectar la taquicardia compensatoria e incrementar el riesgo de hipotensión cuando se usa con anestésicos. El anestesista debe estar informado de que el pacientes está usando COMBIGAN®-D. COMBIGAN®-D contiene cloruro de benzalconio como conservador el cual puede decolorar y ser absorbido por los lentes de contacto suaves; por lo tanto, los pacientes que usan lentes de contacto suaves (hidrofílicos) deben quitarse los lentes antes de la administración de la solución y esperar por lo menos 15 minutos después de la instilación de COMBIGAN®-D antes de colocarse los lentes de contacto suaves. Se les debe indicar a los pacientes que eviten el contacto de la punta del contenedor con el ojo o con estructuras de alrededor para evitar cualquier daño ocular o contaminación de la solución. No hay estudios adecuados y bien controlados de COMBIGAN®-D en niños (menores de 18 años). Ninguna diferencia en cuanto a seguridad y eficacia ha sido observada entre pacientes jóvenes y adultos mayores. COMBIGAN®-D, al igual que otros medicamentos similares, puede causar fatiga y/o somnolencia en algunos pacientes. Pacientes que tengan actividades como manejar y operar maquinaria deben tener precaución debido a la disminución de lucidez mental. Puede ocasionar visión borrosa o disturbios en la visión. Los pacientes deberán esperar a que estos síntomas hayan pasado antes de manejar o usar una máquina.
Restricciones de uso durante el embarazo y la lactancia: No hay estudios adecuados y bien controlados de COMBIGAN®-D en mujeres embarazadas. Debido a que los estudios reproducidos en animales no siempre son pronóstico de la respuesta humana, COMBIGAN®-D debe ser utilizado durante el embarazo sólo si el beneficio potencial a la madre justifica el riesgo potencial al feto. El timolol se ha detectado en la leche materna después de administraciones orales y oftálmicas del medicamento. Los estudios en ratas indican que la brimonidina es excretada en la leche materna. Debido a las reacciones adversas potencialmente serias del infante lactante al timolol o al tartrato de brimonidina, se debe tomar una decisión sobre suspender la lactancia o el medicamento, tomando en consideración la importancia del medicamento para la madre.
Reacciones secundarias y adversas: Las reacciones adversas típicas de los agentes betabloqueadores sistémicos pueden ocurrir debido al componente beta adrenérgico timolol. Raramente se han reportado reacciones respiratorias y cardiacas, incluyendo muerte asociada a insuficiencia cardiaca o debido a broncoespasmos. Se han reportado reacciones tardías de hipersensibilidad ocular con la solución de tartrato de brimonidina al 0.2%, algunos reportes se han asociado con un incremento de la PIO. Durante la post-comercialización se han reportado apnea, bradicardia, coma, hipotensión, hipotermia, hipotonía, letargo, palidez, depresión respiratoria y somnolencia en neonatos, infantes y niños que recibieron brimonidina ya sea por glaucoma congénito o por ingesta accidental. Estudios clínicos: En los estudios clínicos con COMBIGAN®-D, la mayoría de las reacciones fueron transitorias y no de una severidad que requiriera la suspensión del tratamiento. La tabla 1 muestra la información compilada de estudios principales de 12 meses. Las reacciones adversas fueron codificadas usando el diccionario COSTART disponible cuando se hizo el estudio reflejando las reacciones reportadas. La clasificación de los eventos adversos está basada en el Sistema de Clasificación de Órganos MedDRA (SOC).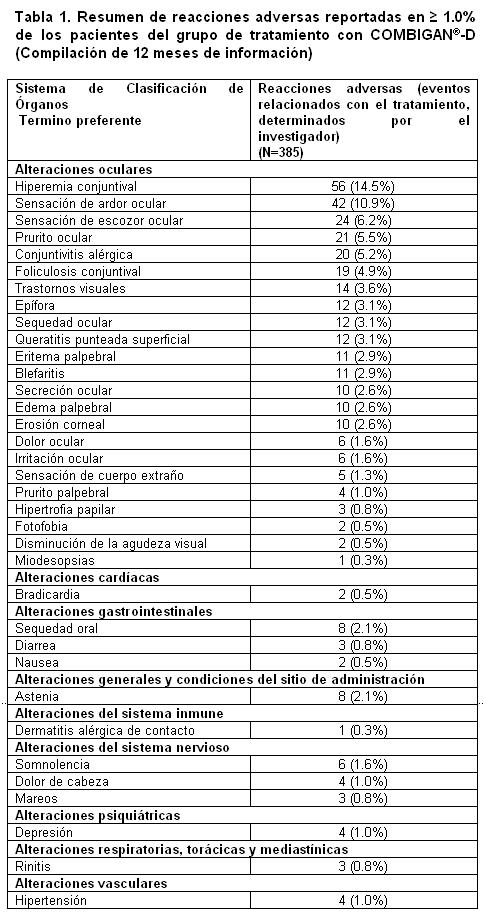
La tabla 2 presenta las reacciones adversas con tasas de incidencia < 1% reportadas en 2 estudios de 12 meses.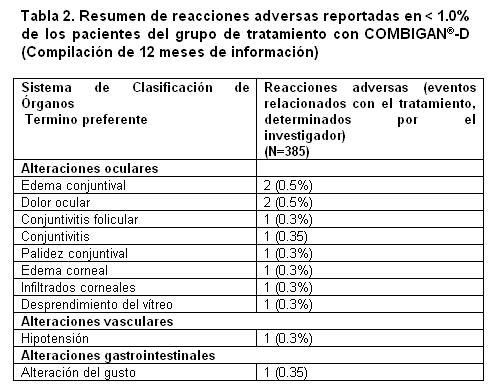
En un estudio de 3 meses fase 3 en niños (edad 2-7 años) con glaucoma inadecuadamente controlado mediante beta-bloqueadores, se reportó una alta prevalencia de somnolencia (55%) con la administración de una solución oftálmica de tartrato de brimonidina al 0.2% como tratamiento adyuvante a beta-bloqueadores tópicos, la somnolencia fue severa en el 8% de los niños por lo que se suspendió el tratamiento en el 13%. La incidencia de somnolencia disminuyó a mayor de edad, siendo el grupo de 7 años el menos afectado por este factor (25%), pero se vio afectado por el peso, presentándose más frecuentemente en los niños con un peso de ≤20kg (63%) en comparación con los niños de un peso de > 20kg (25%).
Reacciones adversas adicionales: Las reacciones adversas adicionales que se han reportado en la experiencia clínica con los componentes individuales se enlistan a continuación: Tartrato de brimonidina: Alteraciones oculares: Palidez conjuntival, edema conjuntival, iritis, iridociclitis (uveítis anterior) y miosis. Alteraciones del sistema inmune: hipersensibilidad, reacción cutánea (incluyendo eritema, edema facial, prurito, erupción cutánea) y vasodilatación. Alteraciones psiquiátricas: Insomnio. Alteraciones cardíacas: palpitaciones / arritmias, taquicardia. Alteraciones vasculares: síncope. Alteraciones respiratorias, torácicas y mediastínicas: sequedad nasal, síntomas en vías respiratorias superiores. Alteraciones gastrointestinales: alteración del gusto y síntomas gastrointestinales. Timolol (administración oftálmica): Alteraciones oculares (también conocidas como de sentidos especiales): desprendimiento coroideo después de una cirugía de filtración, conjuntivitis, edema macular cistoide, disminución de la sensibilidad corneal, diplopía, queratitis, pseudopemfigoide, ptosis y alteraciones de la refracción. Alteraciones cardiacas (también conocido como cardiovasculares): arritmia, bloqueo aurículoventricular, paro cardíaco, insuficiencia cardiaca, dolor de pecho, insuficiencia cardiaca congestiva, edema, bloqueo cardiaco, palpitaciones, edema pulmonar y agravamiento de la angina de pecho. Alteraciones auditivas y laberínticas (también conocidos como de sentidos especiales): Tinnitus. Alteraciones gastrointestinales(también conocidos como digestivos): dolor abdominal, anorexia, disgeusia, dispepsia y vómito. Alteraciones del sistema inmune (también conocidos como de hipersensibilidad o inmunológicos): reacciones alérgicas sistémicas, incluyendo anafilaxia, angioedema, erupción cutánea localizada y generalizada, prurito, urticaria y lupus sistémico eritematoso. Alteraciones nutricionales y metabólicas (también conocidos como endocrinos): hipoglucemia (en pacientes diabéticos). Alteraciones del tejido conectivo y del músculo-esquelético: mialgia. Alteraciones del sistema nervioso: isquemia cerebral, accidente cerebro vascular, incrementos en signos y síntomas de miastenia gravis, parestesia y síncope. Alteraciones Psiquiátricas (también conocidos como del sistema nervioso/psiquiátricos): cambios de comportamiento y trastornos psíquicos incluyendo ansiedad, confusión, desorientación, alucinaciones, insomnio, pérdida de memoria, nerviosismo y pesadillas. Alteraciones del sistema reproductivo y de la mama (también llamados eurogenitales): disminución de la libido, trastorno de Peyronie, fibrosis retroperitoneal, impotencia sexual. Alteraciones respiratorias, torácicas y mediastínicas (también llamados respiratorios): broncoespasmos (predominantemente en pacientes con enfermedad broncoespástica preexistente), tos, disnea, congestión nasal, falla respiratoria e infección en las vías respiratorias superiores. Alteraciones del tejido subcutáneo y piel: alopecia, exacerbación de la psoriasis, erupción psoriasiforme y erupción cutánea. Alteraciones vasculares (tomados parcialmente de los cardiovasculares): claudicación, manos y pies fríos, hipotensión y fenómeno de Raynaud. Experiencia post-marketing: Las siguientes reacciones adversas han sido identificados durante el periodo post-comercialización de COMBIGAN®-D en la práctica clínica, la estimación de la frecuencia no se puede realizar ya que fueron reportados voluntariamente por una población de tamaño desconocido. Alteraciones oculares: visión borrosa, reducción de agudeza visual. Alteraciones del sistema nervioso: mareos. Alteraciones cardiacos: bradicardia. Alteraciones vasculares: hipotensión. Alteraciones gastrointestinales: náusea. Alteraciones del tejido subcutáneo y piel: eritema facial.
Interacciones medicamentosas y de otro género: No se han llevado a cabo estudios de interacción medicamentosa con COMBIGAN®-D. Depresores SNC: Aunque no se han llevado a cabo estudios de interacción medicamentosa con COMBIGAN®-D, se debe considerar la posibilidad de un efecto aditivo o potencializador con depresores del sistema nervioso central (alcohol, barbitúricos, opioides, sedantes o anestésicos). Anti depresivos tricíclicos: Se ha reportado que los antidepresivos tricíclicos bloquean el efecto hipotensión de la clonidina sistémica. No se sabe si el uso concomitante de estos agentes con COMBIGAN®-D en humanos pueda interferir con el efecto reductor de la PIO. No hay información disponible sobre el nivel de catecolaminas circulantes después de la administración de COMBIGAN®-D. Sin embargo, se recomienda tener cuidado con pacientes que están tomando anti depresivos triciclicos los cuales pueden afectar el metabolismo y la absorción de aminas circulantes. Está contraindicada la administración concomitante de inhibidores de la MAO. Los pacientes que han estado recibiendo terapia de inhibición de la MAO deben esperar 14 días después de la suspensión antes de comenzar la terapia con COMBIGAN®-D. Agentes bloqueadores beta adrenérgicos: los pacientes que están recibiendo un agente bloqueador beta-adrenérgico sistémico (ejemplo: vía oral o intravenoso) y COMBIGAN®-D deben ser observados ya que se puede potenciar el efecto aditivo del beta-bloqueador, tanto en la presión sistémica como en la intraocular. Anti-hipertensión / Glucósidos cardiacos: Hay un efecto aditivo potencial que provoca hipotensión, y/o una marcada bradicardia cuando las gotas oftálmicas de beta-bloqueadores son administrados concomitantemente con bloqueadores del canal de calcio orales, anti-arritmias (incluyendo amiodarona), glucósidos digitálicos, parasimpaticomiméticos, guanetidina y otros anti-hipertensivos. Agentes midriáticos: Aunque el timolol tenga poco o nulo efecto sobre el tamaño de la pupila, se ha reportado ocasionalmente midriasis cuando el timolol se ha usado con agentes midriáticos como la adrenalina. Inhibidores del CYP2D6: Se ha reportado una potencialización del beta bloqueo sistémico (ejemplo: decremento de la frecuencia cardiaca, depresión) durante el tratamiento combinado con inhibidores del CYP2D6 [(ejemplo quinidina, inhibidores selectivos de la re captación de serotonina (SSRIs)] y timolol.
Alteraciones en los resultados de pruebas de laboratorio: No se han reportado.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Con tartrato de brimonidina no se observaron efectos carcinogénicos relacionados al compuesto tanto en ratas como en ratones después de un estudio de 21meses y de 24 meses, respectivamente. En estos estudios, la dosis de administración, en la dieta, de tartrato de brimonidina de hasta 2.5 mg/kg/día en ratones y 1.0 mg/kg/día en ratas alcanzó 150 y 210 veces, respectivamente, la concentración máxima plasmática (C max) en humanos tratados con una gota de COMBIGAN®-D en ambos ojos por día. En un estudio a 2 años, de maleato de timolol administrado por vía oral a ratas, hubo un incremento estadísticamente significativo en la incidencia de feocromocitomas suprarrenales en ratas macho a la dosis de 300 mg/kg/día (aproximadamente 25,000 veces la exposición sistémica después de administrar la dosis oftálmica diaria recomendada para humanos). Ni el tartrato de brimonidina ni el maleato de timolol se consideran un peligro genotóxico, de acuerdo a los resultados de un conjunto de pruebas de genotoxicidad. El tartrato de brimonidina no fue mutagénico o clastogénico en una serie de estudios realizados in vitro e in vivo, incluyendo la prueba Ames de reversión bacteriana, ensayo de aberración cromosómica en células de Ovario de Hámster Chino y tres estudios in vivo en ratones CD-1: ensayo huésped-mediado, estudio citogénico y ensayo letal dominante. Por otra parte el maleato de timolol no presentó potencial mutagénico cuando se probó in vivo (ratón) en la prueba del micronúcleo y en el ensayo citogénico (dosis de hasta 800 mg/kg) e in vitro en el ensayo de transformación de células neoplásicas (hasta 100 mg/ml). El tartrato de brimonidina no mostró ser teratogénico al ser administrado oralmente a ratas en gestación durante los días 6 a 15, y durante los días 6 a 18 en conejos. Las dosis más altas de tartrato de brimonidina en ratas (1.65 mg base/kg/día) y en conejos (3.33 mg base/kg/día) alcanzaron valores diarios de exposición en el área bajo la curva de 580 y 37 veces mayores, respectivamente, que los valores similares estimados en humanos tratados con COMBIGAN®-D con una dosis de 1 gota en ambos ojos 2 veces al día. Después de una dosis oral de 14C-brimonidina en ratas gestantes, la brimonidina atravesó la placenta y entro a la circulación fetal teniendo un alcance limitado, produciendo concentraciones sanguíneas fetales de 14C-brimonidina del 10-27% de la concentración de la misma en sangre materna. La brimonidina se depositó predominantemente en la placenta, útero e hígado fetal pero no en el hígado materno. Los estudios de teratogenicidad con timolol en ratones, ratas y conejos a dosis orales de hasta 50 mg/kg/día (que equivalen a 4,200 veces la exposición sistémica después de la dosis oftálmica diaria recomendada en humanos), no demostraron evidencia de malformaciones fetales. Aunque se observó un retraso en la osificación no se observaron efectos adversos en el desarrollo post-natal de las crías. Las dosis de 1,000 mg/kg/día a (83,000 veces la exposición sistémica después de la administración de la dosis oftálmica diaria recomendada en humanos) fueron materno-tóxicos en ratones y provocaron un incremento en el número de resorciones fetales. Esta misma observación se encontró en conejos a dosis de 18,300 veces la exposición sistémica después de la administración de la dosis oftálmica diaria, en este caso sin toxicidad materna aparente. No hubo daño en la fertilidad y reproducción en ratas macho cuando se trataron por 70 días previos al apareamiento y en ratas hembra cuando se trataron por 14 días previos al apareamiento, continuando durante la gestación y lactancia con dosis orales de tartrato de brimonidina. A pesar de que no se midieron las concentraciones sanguíneas de brimonidina en este estudio, se estima que la dosis más alta de tartrato de brimonidina (0.66 mg/kg/día) alcanzó valores de exposición diaria en el área bajo de la curva de 130 veces la vista en humanos tratados con una gota de COMBIGAN®-D en ambos ojos 2 veces al día. Los estudios de reproducción y fertilidad con timolol en ratas no demostraron efectos adversos en la fertilidad en machos o hembras a dosis de hasta 4,200 veces la exposición sistémica después de administrar la dosis máxima oftálmica recomendada en humanos de timolol al 0.5%.
Dosis y vía de administración: Vía de administración.- oftálmica. COMBIGAN®-D se administra de forma tópica en el ojo. La dosis recomendada para COMBIGAN®-D es una gota en el (os) ojo (s) afectado (s) 2 veces al día cada 12 horas. Al igual que con cualquier gota, para reducir la posible absorción sistémica, se recomienda comprimir el saco lagrimal en el canto medio (ocluyendo el punto lagrimal) durante al menos 1 minuto. Esto se debe realizar inmediatamente después de la instilación de cada gota. Si se va a utilizar más de un producto oftálmico, los diferentes medicamentos se deben instilar por lo menos con cinco minutos de intervalo.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: Existe poca información disponible acerca de la sobredosificación en humanos. Se ha reportado bradicardia asociada con el uso de dosis más altas a la recomendada. El tratamiento para tratar una sobredosificación incluye terapia de soporte y sintomática, se debe de mantener la vía aérea permeable. Se han reportado casos de sobredosificación accidental con solución oftálmica de timolol dando como resultado efectos sistémicos similares a los que se presentan con los agentes bloqueadores beta adrenérgicos tales como mareos, dolor de cabeza, dificultad para respirar, bradicardia, hipotensión, bronco espasmo y paro cardiaco. En un estudio in vitro de hemodiálisis usando 14C timolol adicionado a plasma humano o a sangre entera mostró que el timolol fue dializado facilmente de estos fluidos, sin embargo, en un estudio en pacientes con insuficiencia renal mostro que el timolol no fue dializado fácilmente. Sobredosificación con solución oftálmica al 0.2% de tartrato de brimonidina: En los casos recibidos, los eventos reportados ya han sido enlistados en las reacciones adversas. Sobredosificación sistémica por ingesta accidental de solución oftálmica al 0.2% de tartrato de brimonidina: Existe poca información sobre la ingesta accidental de brimonidina en adultos. El único evento adverso reportado hasta la fecha fue hipotensión.
Presentación(es): Caja de cartón con frasco gotero con 5ml y 10 ml.
Recomendaciones sobre almacenamiento: Consérvese a no más de 25°C. Protéjase de la luz.
Leyendas de protección: Manténgase bien cerrado. Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. No se administre a menores de 18 años. El uso durante el embarazo y lactancia, queda bajo responsabilidad del médico.
Nombre y domicilio del laboratorio: Hecho en EUA por: Allergan Sales, LLC. Waco, TX 76712, EUA. Hecho en Brasil por: Allergan Produtos Farmacêuticos Ltda. Av. Guarulhos, 3272, Ponte Grande, Guarulhos- São Paulo- Brasil. Importado y distribuido por: Allergan, S.A. de C.V. Carlos J. Meneses No. 206, Col. Buenavista, C.P. 06350, Deleg. Cuauhtémoc, D.F., México.
Número de registro del medicamento: 627M2005 SSA IV