COMBODART®
GSK
Denominacion genérica: Dutasterida/Tamsulosina.
Forma farmacéutica y formulación: Cápsulas. Cada cápsula contiene: Dutasterida 0.5 mg. Clorhidrato de Tamsulosina 0.4 mg. Excipiente c.b.p. 1 cápsula.
Indicaciones terapéuticas: COMBODART® trata y previene la progresión de la hiperplasia prostática benigna (HPB, del inglés benign prostatic hyperplasia), mediante el alivio de los síntomas, la reducción del tamaño de la próstata (volumen), el mejoramiento de la velocidad de flujo urinario y la reducción del riesgo de retención urinaria aguda (AUR, del inglés acute urinary retention), así como la necesidad de cirugía relacionada con la HPB.
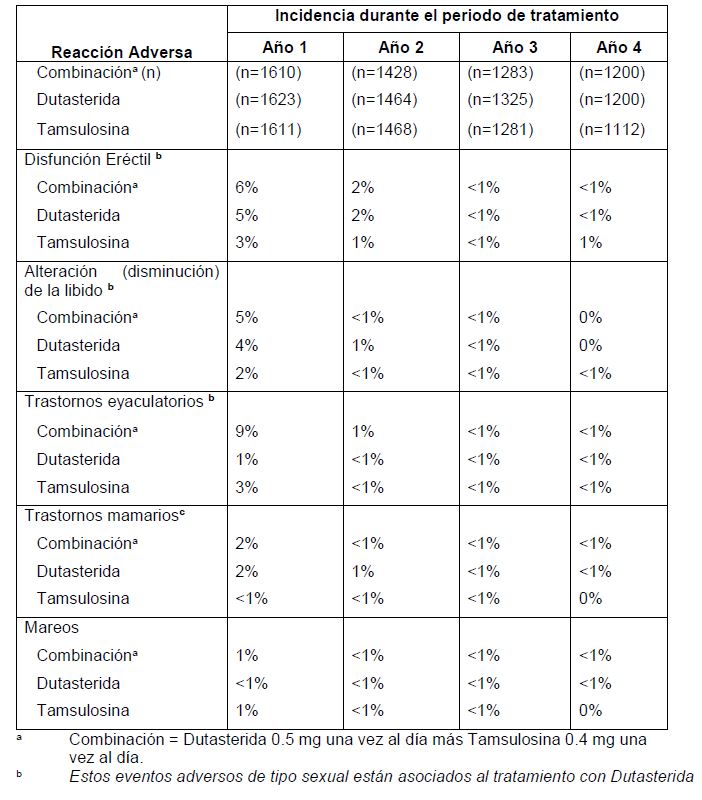
Farmacocinética y farmacodinamia: Farmacocinética: Se demostró la bioequivalencia entre la combinación dutasterida-tamsulosina y la dosificación concomitante con cápsulas separadas de dutasterida y tamsulosina. El estudio sobre bioequivalencia con dosis únicas fue realizado en estados tanto de ayuno como postprandial. Para el componente Tamsulosina de dutasterida- tamsulosina, se observó una reducción de 30% en la Cmáx en el estado postprandial, en comparación con el estado de ayuno. La ingestión de alimentos no produjo efecto alguno en el AUC de la Tamsulosina. Absorción: Dutasterida: La Dutasterida se administra vía oral en solución, como una cápsula de gelatina blanda. Después de la administración de una dosis única de 0.5 mg, se producen concentraciones séricas máximas de Dutasterida en un lapso de 1 a 3 horas. La biodisponibilidad absoluta en el hombre es de aproximadamente 60%, en relación con una infusión intravenosa de 2 horas. La biodisponibilidad de la Dutasterida no se ve afectada por la ingestión de alimentos. Tamsulosina: El clorhidrato de Tamsulosina es absorbido de los intestinos y tiene un nivel de biodisponibilidad casi completo. Después de administrar dosis únicas y múltiples, el clorhidrato de Tamsulosina exhibe un perfil cinético lineal, alcanzando concentraciones en estado estacionario al quinto día de un régimen posológico de una vez al día. La velocidad de absorción del clorhidrato de Tamsulosina se ve reducida con la recién ingestión de alimentos. El paciente puede promover la uniformidad en el nivel absorción si siempre toma su dosis de clorhidrato de Tamsulosina aproximadamente 30 minutos después de la misma comida, cada día. Distribución: Dutasterida: Los datos farmacocinéticos obtenidos después de administrar dosis orales únicas y repetidas, muestran que la dutasterida tiene un gran volumen de distribución (300 a 500 L). La Dutasterida exhibe un alto grado de fijación a proteínas plasmáticas ( > 99.5%). Después de la dosificación diaria, las concentraciones séricas de Dutasterida alcanzan un 65% de la concentración en estado estacionario después de 1 mes, y aproximadamente 90% después de 3 meses. Después de 6 meses de dosificación de 0.5 mg una vez al día, se alcanzan concentraciones séricas en estado estacionario (Css) de aproximadamente 40 ng/mL. En forma similar a las séricas, las concentraciones seminales de Dutasterida alcanzaron el estado estacionario a los 6 meses. Después de 52 semanas de terapia, las concentraciones seminales de Dutasterida promediaron 3.4 ng/mL (intervalo de 0.4 a 14 ng/mL). La repartición de Dutasterida del suero al semen promedió 11.5%. Tamsulosina: La media del volumen aparente de distribución en estado estacionario del clorhidrato de Tamsulosina, después de su administración intravenosa a diez hombres adultos sanos, fue de 16L, lo cual sugiere que existe distribución en los líquidos extracelulares del organismo. El clorhidrato de Tamsulosina tiene un amplio grado de fijación a proteínas plasmáticas humanas (94% a 99%), principalmente a la glucoproteína de ácido alfa-1 (AAG), con una fijación lineal a lo largo de un amplio intervalo de concentraciones (20 a 600 ng/mL). Biotransformación: Dutasterida: In vitro, la Dutasterida se metaboliza, a través de la isoenzima CYP3A4 del citocromo P450 humano, a dos metabolitos monohidroxilados secundarios, pero no se metaboliza a través de las isoenzimas CYP1A2, CY2A6, CYP2E1, CYP2C8, CYP2C9, CYP2C19, CYP2B6 o CYP2D6. Después de la dosificación hasta el estado estacionario, se han detectado en el suero humano Dutasterida inalterada, 3 metabolitos principales (4'-hidroxidutasterida, 1,2- dihidrodutasterida y 6-hidroxidutasterida) y 2 metabolitos secundarios (6,4'- dihidroxidutasterida y 15-hidroxidutasterida), evaluados por respuesta espectrométrica de masa. Los cinco metabolitos séricos humanos de la Dutasterida han sido detectados en el suero de la rata, aunque se desconoce la estereoquímica de las adiciones de hidroxilo en las posiciones 6 y 15 en los metabolitos del ser humano y de la rata. Tamsulosina: No existe bioconversión enantiomérica de clorhidrato de Tamsulosina [R(-) isómero] al S(+) isómero en seres humanos. El clorhidrato de Tamsulosina se metaboliza ampliamente a través de la enzimas del citocromo P450 en el hígado, y menos de 10% de la dosis se excreta de manera inalterada en la orina. Sin embargo, no se ha establecido el perfil farmacocinético de los metabolitos en seres humanos. Los resultados de estudios realizados in vitro indican que las isoenzimas CYP3A4 y CYP2D6 se encuentran implicadas en el metabolismo de la Tamsulosina, con una menor participación de otras isoenzimas CYP. La inhibición de enzimas hepáticas metabolizantes de fármacos podría ocasionar un incremento en el nivel de exposición a la tamsulosina. (Ver Precauciones Generales e Interacciones Medicamentosas y de otro género). Los metabolitos del clorhidrato de Tamsulosina experimentan una amplia conjugación a glucurónido o sulfato, antes de su excreción renal. Eliminación: Dutasterida: La Dutasterida experimenta un amplio metabolismo. Después de la dosificación oral de 0.5 mg/día de dutasterida hasta alcanzar el estado estacionario en seres humanos, de 1.0% a 15.4% (media de 5.4%) de la dosis administrada se excreta como Dutasterida en las heces. El resto se excreta en las heces como 4 metabolitos principales, constituyendo cada uno 39%, 21%, 7% y 7% del material relacionado con el fármaco, y como 6 metabolitos secundarios (menos de 5% cada uno). En la orina humana sólo se detectan rastros de Dutasterida inalterada (menos de 0.1% de la dosis). A concentraciones terapéuticas, la vida media terminal de la Dutasterida es de 3 a 5 semanas. Las concentraciones séricas permanecen detectables (mayores de 0.1 ng/mL) por un período de hasta 4 a 6 meses después de la suspensión del tratamiento. A concentraciones séricas bajas (menos de 3 ng/mL), la Dutasterida se depura rápidamente por las vías de eliminación tanto dependiente como independiente de la concentración. Las dosis únicas de 5 mg, o menores, mostraron indicios de una rápida depuración y una vida media corta de 3 a 9 días. A concentraciones séricas superiores a 3 ng/mL, la Dutasterida se depura lentamente (0.35 a 0.58 L/h), principalmente por eliminación lineal no saturable, con una vida media terminal de 3 a 5 semanas. A concentraciones terapéuticas, después de la dosificación repetida de 0.5 mg/día, la depuración más lenta es la que domina, y la depuración total es lineal e independiente de la concentración. Tamsulosina: La vida media de la tamsulosina es de 5 a 7 h. Aproximadamente 10% se excreta de manera inalterada en la orina. Personas de edad avanzada: Dutasterida: Los perfiles farmacocinético y farmacodinámico de la Dutasterida fueron evaluados en 36 hombres sanos, de 24 a 87 años de edad, después de la administración de una dosis única de 5 mg de Dutasterida. El nivel de exposición a la Dutasterida, representado por valores de AUC y Cmáx, no fue estadísticamente distinto cuando se compararon grupos de edad. La vida media no fue estadísticamente distinta al comparar el grupo de 50-69 años de edad con el grupo de pacientes mayores de 70 años de edad, intervalo de edad con la mayor cantidad de hombres con HPB. Entre los grupos de edad, no se observaron diferencias en el efecto del fármaco, según la medición de la reducción en la concentración de DHT. Los resultados indicaron que no es necesario realizar ajustar la dosis de Dutasterida con base en la edad. Tamsulosina: La comparación de estudios entrecruzados de la exposición general (AUC) y la vida media del clorhidrato de Tamsulosina indican que la eliminación farmacocinética del clorhidrato de Tamsulosina podría verse ligeramente prolongada en hombres de edad avanzada, en comparación con hombres voluntarios sanos y jóvenes. La depuración intrínseca es independiente del grado de fijación del clorhidrato de Tamsulosina a la AAG, pero disminuye con la edad, dando como resultado un nivel de exposición general (AUC) 40% mayor en sujetos de 55 a 75 años de edad, en comparación con sujetos de 20 a 32 años de edad. Insuficiencia renal: Dutasterida: No se ha estudiado el efecto que produce la presencia de insuficiencia renal en el perfil farmacocinético de la Dutasterida. Sin embargo, menos de 0.1% de una dosis de 0.5 mg de Dutasterida en estado estacionario se recupera en la orina humana, por lo cual no se anticipa ajuste alguno en la dosificación de los pacientes con insuficiencia renal. Tamsulosina: El perfil farmacocinético del clorhidrato de Tamsulosina ha sido comparado en 6 sujetos con insuficiencia renal leve-moderada (30 ≤ CLcr < 70 mL/min/1.73m2) o moderada- severa (10 ≤ CLcr < 30 mL/min/1.73m2), y 6 sujetos normales (CLcr > 90 mL/min/1.73m2). Como resultado de la alteración en el grado de fijación a la AAG, se observó un cambio en la concentración plasmática general de clorhidrato de Tamsulosina. No obstante, la concentración libre (activa) de clorhidrato de Tamsulosina, así como la depuración intrínseca, permanecieron relativamente constantes. Por lo tanto, los pacientes que padecen insuficiencia renal no requieren ajuste alguno en su dosificación de clorhidrato de Tamsulosina en cápsulas. Sin embargo, no se han realizado estudios en pacientes con enfermedad renal en etapa terminal (CLcr < 10 mL/min/1.73m2). Insuficiencia hepática: Dutasterida: No se ha estudiado el efecto que produce la presencia de insuficiencia hepática en el perfil farmacocinético de la Dutasterida (Ver Precauciones Generales). Como la Dutasterida experimenta un amplio metabolismo, es posible que el nivel de exposición a este fármaco sea mayor en aquellos pacientes con deterioro hepático. Tamsulosina: El perfil farmacocinético del clorhidrato de Tamsulosina ha sido comparado en 8 sujetos con disfunción hepática moderada (clasificación de Child-Pugh: Grados A y B) y 8 sujetos normales. Como resultado de la alteración en el grado de fijación a la AAG, se observó un cambio en la concentración plasmática general de clorhidrato de Tamsulosina. No obstante, la concentración libre (activa) de clorhidrato de Tamsulosina no cambia significativamente y sólo se observa un cambio modesto (32%) en la depuración intrínseca del clorhidrato de Tamsulosina libre. Por lo tanto, los pacientes que padecen disfunción hepática moderada no requieren ajuste alguno en su dosificación de clorhidrato de Tamsulosina. El clorhidrato de Tamsulosina no ha sido estudiado en pacientes con disfunción hepática severa. Farmacodinamia: COMBODART® es una combinación de dos fármacos, con mecanismos de acción complementarios, para mejorar los síntomas en pacientes con HPB: Dutasterida, un inhibidor dual de la 5 a-reductasa (5 ARI, del inglés 5 a-reductase inhibitor) y clorhidrato de Tamsulosina, un antagonista de los receptores adrenérgicos a1a. El perfil farmacodinámico de COMBODART®; como combinación fija no se esperaría que los efectos fueran diferentes de aquellos producidos por la Dutasterida y la Tamsulosina co-administradas como componentes separados. Dutasterida: La Dutasterida es un inhibidor dual de la 5 alfa-reductasa. Inhibe a las isoenzimas tipo 1 y tipo 2 de la 5 alfa-reductasa, las cuales son responsables de la conversión de testosterona a 5 alfa-dihidrotestosterona (DHT). La DHT es el andrógeno responsable principalmente de la hiperplasia del tejido glandular prostático. La Dutasterida disminuye las concentraciones de DHT, reduce el volumen prostático, mejora los síntomas de las vías urinarias inferiores y el flujo urinario, y reduce el riesgo de RUA o cirugía relacionada con HPB. El efecto máximo de las dosis diarias de Dutasterida en la reducción de la concentración de DHT depende de la dosis y se observa dentro de 1 a 2 semanas. Después de 1 semana y 2 semanas de dosificación diaria de 0.5 mg de Dutasterida, las concentraciones séricas medias de DHT se vieron reducidas en 85% y 90%, respectivamente. En pacientes con HPB tratados con 0.5 mg de Dutasterida al día, la reducción mediana en la concentración de DHT fue de 94% a 1 año y de 93% a 2 años; asimismo, el incremento mediano en la concentración sérica de testosterona fue de 19%, tanto a 1 y 2 años. Esta es una consecuencia esperada de la inhibición de la 5 alfa-reductasa, la cual no dio lugar a eventos adversos conocidos. Tamsulosina: La Tamsulosina inhibe los receptores adrenérgicos a1a en el músculo liso del estroma prostático y el cuello de la vejiga. Aproximadamente el 75% de los receptores prostáticos a1 son del subtipo a1a. La Tamsulosina incrementa la velocidad de flujo urinario máximo al reducir la tensión del músculo liso en la próstata y la uretra, liberando así la obstrucción. También mejora el complejo de síntomas irritativos y obstructivos en los que desempeñan un papel importante la inestabilidad de la vejiga y la tensión de los músculos lisos de las vías urinarias inferiores. Los bloqueadores adrenérgicos alfa-1 son capaces de reducir la presión arterial al disminuir la resistencia periférica. Estudios Clínicos: Las siguientes declaraciones reflejan la información disponible sobre la Dutasterida y la Tamsulosina administradas de manera separada o como terapia de combinación. En un estudio multicéntrico, doble ciego, y realizado en grupos paralelos durante 2 años, se evaluó la administración de 0.5mg/día de AVODART®, 0.4mg/día de Tamsulosina, o la coadministración de 0.5mg de AVODART® más 0.4mg de Tamsulosina, en 4844 hombres con próstata hipertrofiada (mayor o igual a 30cc). El criterio principal de valoración de eficacia a los 4 años de tratamiento fue el nivel de mejoría, con respecto a la línea basal, en la calificación internacional de síntomas prostáticos (IPSS, del inglés international prostate symptom score). La combinación de Dutasterida y Tamsulosina proporciona una mejoría en los síntomas superior a la de cualquier componente solo. Después de 2 años de tratamiento, la terapia de combinación demostró una mejoría media ajustada estadísticamente significativa en las calificaciones de síntomas con respecto a la línea basal, de -6.2 unidades. Las mejorías medias ajustadas en las calificaciones de síntomas, observadas con las terapias individuales, fueron de -4.9 unidades para AVODART® y de -4.3 unidades para la Tamsulosina. La mejoría media ajustada en la velocidad de flujo con respecto a la línea basal fue de 2.4 ml/seg para la combinación, 1.9 ml/seg para AVODART® y 0.9 ml/seg para la Tamsulosina. La mejoría media ajustada en el Índice de Impacto de la HPB (BII del inglés HPB Impact Index), con respecto a la línea basal, fue de -2.1 unidades para la combinación, -1.7 para AVODART® y -1.5 para la Tamsulosina. Estas mejorías en la velocidad de flujo y BII fueron estadísticamente significativas para la terapia de combinación, en comparación con ambas monoterapias. Después de 2 años de tratamiento, la reducción en el volumen prostático total y el volumen de la zona de transición fue estadísticamente significativa para la terapia de combinación, en comparación con la monoterapia con Tamsulosina. El criterio primario de valoración de eficacia a los 4 años de tratamiento fue el tiempo hasta el primer evento de RUA o cirugía relacionada con HPB. Después de 4 años de tratamiento, la terapia combinada redujo de una manera estadísticamente significativa el riesgo de desarrollar AUR o necesitar una cirugía relacionada con HPB (reducción de 65.8% en el riesgo, p < 0.001 [IC de 95% 54.7% to 74.1%]), en comparación con la monoterapia con tamsulosina. La incidencia de AUR o cirugía relacionada con HPB hacia el año 4 fue de 4.2% para la terapia de combinación y de 11.9% para la tamsulosina (p < 0.001). En comparación con la monoterapia con AVODART®, la terapia combinada redujo el riesgo de desarrollar AUR o necesitar una cirugía relacionada con HPB en 19.6%; la diferencia observada entre los grupos de tratamiento no fue significativa (p=0.18 [IC de 95% -10.9% to 41.7%]). La incidencia de AUR o cirugía relacionada con HPB hacia el año 4 fue de 4.2% para la terapia combinada y de 5.2% para AVODART®. La progresión clínica se definió como un término compuesto de empeoramiento de los síntomas, (IPSS), eventos de RUA relacionados con HPB, incontinencia, UTI e insuficiencia renal. La terapia combinada estuvo asociada con una disminución estadísticamente significativa en la tasa de progresión clínica, en comparación con la tamsulosina (p < 0.001, reducción del riesgo de 44.1% [IC de 95 %: 33.6% a 53.0%]), después de 4 años. Las tasas de progresión clínica para la terapia de combinación, la tamsulosina y AVODART® fueron: 12.6%, 21.5% y 17.8%, respectivamente. Del año 2 al año 4, se mantuvo la media ajustada de la mejoría estadísticamente significativa en las puntuaciones de los síntomas (IPSS), con respecto a la línea basal. A los 4 años, las medias ajustadas de las mejorías observadas en las puntuaciones de los síntomas fueron de -6.3 unidades para la terapia de combinación, -5.3 unidades para la monoterapia con AVODART® y -3.8 unidades para la monoterapia con tamsulosina. Después de 4 años de tratamiento, la media ajustada de la mejoría en la velocidad de flujo (Qmax), con respecto a la línea basal, fue de 2.4 ml/seg para la terapia de combinación, 2.0 ml/seg para la monoterapia con AVODART® y 0.7 ml/seg para la monoterapia con tamsulosina. En comparación con la tamsulosina, la media ajustada de la mejoría en la Qmax con respecto a la línea basal fue mayor (de una manera estadísticamente significativa) con la terapia de combinación en cada evaluación semestral, del Mes 6 al Mes 48 (p < 0.001). En comparación con AVODART®, la media ajustada de la mejoría en la Qmax con respecto a la línea basal no difirió de una manera estadísticamente significativa con aquella observada con la terapia de combinación (p=0.050 en el Mes 48). La terapia de combinación fue significativamente superior (p < 0.001) a la monoterapia con tamsulosina y a la monoterapia con AVODART®, en cuanto a la mejoría en los parámetros de desenlaces de salud BII y el Estado de Salud relacionado con la HPB (BHS, HPB-related Health Status) a los 4 años. La media ajustada de la mejoría en los BII con respecto a la línea basal fue de -2.2 unidades para la combinación, -1.8 para AVODART® y -1.2 para la tamsulosina. La media ajustada de la mejoría en el BHS con respecto a la línea basal fue de -1.5 unidades para la combinación, -1.3 para AVODART® y -1.1 para la tamsulosina. Después de 4 años de tratamiento, la reducción en el volumen total de la próstata y el volumen de la zona de transición fue estadísticamente significativa para la terapia de combinación, en comparación con la monoterapia con tamsulosina sola. Dutasterida: Se evaluó la administración de dutasterida 0.5 mg/día o placebo en 4325 hombres con próstatas hipertrofiadas (mayores de 30 cc) en tres estudios doble ciego, multicéntricos, controlados con placebo y de 2 años de duración para evaluar la eficacia primaria. En hombres con HPB, la dutasterida trata y previene la progresión de la enfermedad reduciendo tanto el riesgo de desarrollar RUA (retención urinaria aguda) así como la necesidad de intervención quirúrgica, y suministrando una mejoría estadísticamente significativa de los síntomas del tracto urinario inferior (LUTS, del inglés lower urinary tract symptoms), la velocidad de flujo urinario máximo (Qmax) y el volumen prostático, en comparación con el placebo. Estas mejorías en los LUTS, el Qmax y el volumen prostático se observaron a lo largo de 24 meses, y los LUTS y el Qmax siguieron mejorando durante un periodo adicional de 2 años, en estudios de extensión abierta. Además, las reducciones en el volumen prostático fueron constantes durante un periodo adicional de 2 años, en estudios de extensión abierta. Insuficiencia cardiaca: En un estudio de 4 años de comparación de AVODART® co-administrado con tamsulosina y AVODART® o monoterapia de tamsulosina en hombres con HPB (el estudio CombAT) la incidencia del término compuesto de insuficiencia cardiaca en el grupo de combinación (14/1610, 0.9%) fue mayor que en el grupo de monoterapia: AVODART®, 4/1623 (0.2%) y tamsulosina, 10/1611, (0.6%). El valor estimado de riesgo relativo para el tiempo al primer evento de insuficiencia cardiaca fue de 3.57 [IC de 95% 1.17, 10.8], para el tratamiento de combinación comparado con la monoterapia con AVODART®, y de 1.36 [IC de 95% 0.61, 3.07] comparado con la monoterapia con tamsulosina. En el estudio REDUCE de quimio prevención, en donde se llevo a cabo una comparación de 4 años de placebo y dutasterida en 8,231 hombres de 50 a 75 años de edad, con biopsia previa negativa para cáncer de próstata y APE basal de entre 2.5 ng/mL y 10.0 ng/mL,hubo una mayor incidencia del término compuesto de insuficiencia cardiaca en sujetos que tomaron AVODART® (30/4105, 0.7%) frente a placebo (16/4126, 0.4%), en cuanto al estimador de riesgos relativos para el tiempo al primer evento de insuficiencia cardiaca de 1.91 [IC de 95% 1. 4, 3. 50]. En un análisis post-hoc del uso concomitante de bloqueadores alfa, hubo una mayor incidencia del término compuesto de insuficiencia cardiaca en sujetos que tomaron AVODART® y algún bloqueador alfa de manera concomitante (12/1152, 1.0%), en comparación con los sujetos que no tomaron concomitantemente dutasterida y algún bloqueador alfa: AVODART® y ningún bloqueador alfa (18/2953, 0.6%), placebo y algún bloqueador alfa (1/1399, < 0.1%), placebo y ningún bloqueador alfa (15/2727, 0. 6%). No se ha establecido relación causal alguna entre la administración de dutasterida (sola o en combinación con algún bloqueador alfa) y el desarrollo de insuficiencia cardiaca (Ver Precauciones Generales). Cáncer de próstata y tumores de alto grado: En un estudio de 4 años de comparación de placebo y AVODART® en 8,231 hombres de 50 a 75 años de edad, con biopsia previa negativa para cáncer de próstata y APE (antígeno prostático especifico) basal de entre 2.5 ng/mL y 10.0 ng/mL (el estudio REDUCE), a lo largo de 4 años, 6,706 sujetos tuvieron datos disponibles de biopsias por punción con aguja, a fin de determinar las puntuaciones de Gleason mediante análisis. En el estudio se observaron 1,517 sujetos diagnosticados con cáncer de próstata. En ambos grupos de tratamiento, la mayoría de los cánceres de próstata detectables mediante biopsia fueron diagnosticados como de bajo grado (Gleason 5-6) No hubo diferencia alguna en la incidencia de los cánceres grado 7-10 de Gleason (p=0.81). Se observó una alta incidencia de cánceres de próstata de grado 8-10 de Gleason en el grupo tratado con AVODART® (m=29, 0.9%), en comparación con el grupo placebo (19, 0.6%) (p=0.15). En los Años 1-2, el número de sujetos con cánceres de grado 8-10 de Gleason fue similar en el grupo tratado con AVODART® (n=17, 0.5%) y en el grupo placebo (n=18, 0.5%). En los Años 3-4, se diagnosticaron más cánceres de grado 8-10 de Gleason en el grupo tratado con AVODART® (n=12, 0.5%), en comparación con el grupo placebo (n=1, < 0.1%) (p=0.0035). No existen datos disponibles acerca del efecto de dutasterida más allá de 4 años en hombres con riesgo de desarrollar cáncer de próstata. En el grupo tratado con AVODART®, e l porcentaje de sujetos diagnosticados con cánceres de grado 8-10 de Gleason fue consistente en todos los periodos de tiempo del estudio (Años 1-2 y Años 3-4) (0.5% en cada periodo de tiempo), mientras que en el grupo placebo, el porcentaje de sujetos diagnosticados con cánceres de grado 8-10 de Gleason fue más bajo durante los Años 3-4 que en los Años 1-2 ( < 0.1% frente a 0.5%, respectivamente). En un estudio de HPB de 4 años de duración (CombAT), en el cual no hubo biopsias obligatorias por el protocolo y todos los diagnósticos de cáncer de próstata estuvieron sustentados por biopsias "con causa", las tasas de cáncer Gleason 8-10 fueron de (n=8, 0.5%) para AVODART®, (n=11, 0.7%) para tamsulosina y (n=5, 0.3%) para el tratamiento combinado (Ver Precauciones Generales). Los resultados de un estudio epidemiológico, basado en la población (n=174,895) en centros sanitarios de la comunidad mostró que el uso de 5-ARIs para tratar BPH/LUTS no está asociado a un aumento del riesgo en la mortalidad por cáncer de próstata (cociente de riesgo ajustado para riesgos en competencia: 0.85, 95% IC 0.72, 1.01) comparado con el uso de alfa-bloqueadores. Se reportaron resultados similares en un estudio epidemiológico (n=13,892) de hombres con cáncer de próstata en UK (cociente de riesgo ajustado para mortalidad por cáncer de próstata para usuarios de 5-ARI versus no usuarios: 0.86; 95% IC 0.69, 1.06). Un estudio de cohortes prospectivo, el Estudio de Seguimiento de los Profesionales de la Salud (n=38,058), también mostró que el uso de 5-ARI no estaba asociado con cáncer de próstata fatal (HR ajustado: 0.99; 95% IC 0.58, 1.69). Efectos en el antígeno prostático específico (APE) y detección de cáncer de próstata: En el estudio REDUCE en donde pacientes con biopsia previa negativa para cáncer de próstata y APE basal de entre 2.5 ng/mL y 10.0 ng/mL el tratamiento con AVODART® ocasionó un decremento en los niveles séricos medios de APE en aproximadamente 50% después de seis meses de tratamiento, con una gran variabilidad (desviación estándar de 30%) entre los pacientes. La supresión de APE observada a los seis meses fue similar en hombres que sí desarrollaron o que no desarrollaron cáncer de próstata detectable mediante biopsia durante el estudio (Ver Precauciones Generales). Incidencia de cáncer de mama: En estudios clínicos realizados con monoterapia para el tratamiento de la HPB, los cuales brindaron 3,374 años-paciente de exposición a AVODART®, hubo 2 casos de cáncer de mama en hombres reportados en los pacientes tratados con AVODART®; uno después de 10 semanas de tratamiento y el otro después de 11 meses de tratamiento, así como 1 caso en un paciente que recibió placebo. En estudios clínicos subsiguientes para evaluar la reducción del riesgo de HPB y cáncer de próstata, los cuales brindaron 17489 años-paciente de exposición a AVODART® y 5027 años-paciente de exposición a la combinación de AVODART® y tamsulosina, no hubo casos reportados de cáncer de mama en ninguno de los grupos de tratamiento. Dos estudios epidemiológico de casos y controles, uno conducido en una base de datos de salud en US (n=339 casos de cáncer de mama y n=6,780 controles) y el otro en una base de datos de salud en UK (n=398 casos de cáncer de mama y n=3,930 controles), no mostró aumento en el riesgo de desarrollar cáncer de mama en hombres con el uso de 5-ARIs (véase Advertencias y Precauciones). Los resultados del primer estudio no identificaron una asociación positiva para cáncer de mama en hombres (riesgo relativo por ≥ 1 año de uso antes del diagnóstico de cáncer de mama comparado con < 1 año de uso: 0.70: 95% IC 0.34, 1.45). En el segundo estudio, la proporción de probabilidades estimada para cáncer de mama asociada con el uso de 5-ARIs comparada con no uso fue 1.08: 95% CI 0.62, 1.87). No se ha establecido la relación entre el uso a largo plazo de AVODART® y cáncer de mama masculino.
Contraindicaciones: COMBODART® está contraindicado en pacientes con hipersensibilidad conocida a la dutasterida, a otros inhibidores de la 5 alfa-reductasa, al clorhidrato de tamsulosina o a cualquier componente de la preparación. El uso de COMBODART® está contraindicado en mujeres y niños (Ver Restricciones de Uso durante el Embarazo y la Lactancia).
Precauciones generales: Cáncer de próstata: En un estudio de 4 años con más de 8,000 hombres de 50 a 75 años de edad, con una biopsia previa negativa para cáncer de próstata y un APE basal entre 2.5 ng/mL y 10.0 ng/mL (el estudio REDUCE), 1,517 hombres fueron diagnosticados con cáncer de próstata. Hubo una incidencia más alta de Gleason 8 a 10 de cánceres de próstata en el grupo AVODART®(n=29, 0.9%) comparado con el grupo placebo (n=19, 0.6%). No hubo una incidencia aumentada de Gleason 5-6 ó 7-10 de cánceres de próstata. No se ha establecido que haya una relación causal entre AVODART® y el cáncer de próstata de alto grado. Se desconoce la significancia clínica del desequilibrio numérico. Hombres tomando COMBODART® deben ser evaluados regularmente para el riesgo de cáncer de próstata incluyendo determinación de PSA. En un estudio de seguimiento adicional de 2 años con los pacientes originales del estudio AVODART® de quimioprevención (REDUCE), se diagnosticó un índice bajo de nuevos casos de cáncer de próstata (dutasteride [n=14, 1.2%] y placebo [n=7, 0.7%]), sin que se identificaran nuevos casos de cánceres de próstata Gleason 8-10. En un estudio de seguimiento a largo plazo (hasta 18 años) de quimioprevención de otro 5-ARI (finasteride) no mostró diferencia estadísticamente significativa entre finasteride y placebo en las tasas de sobrevida global (HR 1.02, 95% IC 0.97-1.08) o de sobrevida después de los diagnósticos de cáncer de próstata (HR 1.01, 95% IC 0.85- 1.20). Antígeno Prostático Específico: La concentración de APE es un componente importante del proceso de selección para detectar cáncer de próstata. COMBODART® ocasiona una disminución en las concentraciones medias séricas de APE, de aproximadamente 50% después de 6 meses de tratamiento. Los pacientes que reciben tratamiento con COMBODART® deberán tener un nuevo nivel basal de APE establecido después de 6 meses de tratamiento con COMBODART®. A partir de entonces, se recomienda vigilar periódicamente los niveles de APE. Cualquier incremento sostenido a partir del nivel más bajo de APE mientras se esté bajo tratamiento con COMBODART® podría evidenciar la presencia de cáncer de próstata o una falta de cumplimiento de la terapia con COMBODART®, y debe evaluarse cuidadosamente, aún si esos valores siguen estando en el intervalo normal en hombres que no toman algún inhibidor de la 5-ARI (Ver Farmacocinética y Farmacodinamia, Estudios Clínicos). En la interpretación de un valor del APE de un paciente que esté tomando COMBODART®, deben buscarse los valores previos de APE para comparación. El tratamiento con COMBODART® no interfiere con el uso del APE como una herramienta para ayudar en el diagnóstico de cáncer de próstata, después de haber establecido un nuevo nivel basal. Las concentraciones séricas totales de APE regresan a los valores basales dentro de los 6 meses posteriores a la suspensión del tratamiento. El cociente de APE libre a total permanece constante aún bajo la influencia de COMBODART®. Si los médicos eligen utilizar el porcentaje de APE libre como auxiliar en la detección de cáncer de próstata en hombres sometidos a terapia con COMBODART®, no es necesario ajustar su valor. Los pacientes con BPH deben someterse a una exploración digital rectal, así como a otras evaluaciones de cáncer de próstata, antes de iniciar la terapia con DUODART y periódicamente a partir de entonces. Eventos adversos cardiovasculares: En dos estudios clínicos de 4 años, la incidencia de insuficiencia cardiaca (un término compuesto de eventos reportados, insuficiencia cardiaca principalmente e insuficiencia cardiaca congestiva) fue más alto entre sujetos tomando la combinación de AVODART y un bloqueador alfa, principalmente tamsulosin, que entre sujetos no tomando la combinación. En esos dos estudios, la incidencia de insuficiencia cardiaca fue baja (≤1%) y variable entre los estudios. No se observó desequilibrio en la incidencia global de eventos adversos cardiovasculares en ninguno de los estudios. No se ha establecido que haya relación causal entre AVODART (sólo o en combinación con un bloqueador alfa) e insuficiencia cardiaca (ver Estudios Clínicos). Cáncer de mama: Ha habido raros reportes de cáncer de mama en hombres tomando AVODART en estudios clínicos y durante el período de post-mercadeo. Sin embargo, estudios epidemiológicos no demostraron aumento en el riesgo de desarrollar cáncer de mama en hombres con el uso de 5-ARIs. Los médicos prescriptores deben instruir a sus pacientes para que reporten tempranamente cualquier cambio en su tejido mamario tales como tumoraciones o descarga a través del pezón. Hipotensión: Al igual que con otros bloqueadores adrenérgicos alfa-1, se puede presentar hipotensión ortostática en pacientes tratados con tamsulosina, la cual en raras ocasiones puede producir síncope. A los pacientes que comiencen un tratamiento con COMBODART®, se les debe advertir que se sienten o se recuesten a los primeros signos de hipotensión ortostática (mareos y vértigo), hasta que los síntomas se hayan resuelto. Se aconseja precaución cuando agentes bloqueadores alfa adrenérgicos, incluyendo la tamsulosina, se co-administran con inhibidores de la PDE5. Tanto los bloqueadores alfa adrenérgicos y los inhibidores de la PDE5 son vasodilatadores que pueden disminuir la presión arterial. El uso concomitante de estas dos clases de fármacos puede potencialmente causar hipotensión sintomática (Ver Interacciones Medicamentosas y de Otro Género). Síndrome intraoperatorio de iris flácido: Se han observado casos de síndrome intraoperatorio de iris flácido (IFIS, del inglés Intraoperative Floppy Iris Syndrome; una variante del síndrome de pupila pequeña) durante la cirugía de cataratas en algunos pacientes tratados con bloqueadores adrenérgicos alfa-1, incluyendo tamsulosina. El IFIS podría aumentar el riesgo de complicaciones oculares durante y después de los procedimientos operatorios. Durante la evaluación preoperatoria, los cirujanos de cataratas y los equipos de oftalmología deberán considerar si los pacientes programados para cirugía de cataratas están siendo o han sido tratados con COMBODART®, a fin de garantizar que se tomen las medidas adecuadas para tratar el IFIS, en caso de que se presente durante la cirugía. Se considera anecdóticamente útil la suspensión del tratamiento con tamsulosina 1 - 2 semanas antes de la cirugía de cataratas, pero aún no se establece el beneficio ni la duración de la suspensión de la terapia, antes de la cirugía de cataratas. Cápsulas que presenten derrames: La Dutasterida se absorbe a través de la piel, por lo que mujeres y niños deben evitar el contacto con cápsulas que presenten derrames. Si hubiera contacto con cápsulas con fuga, el área debe ser lavada inmediatamente con jabón y agua (véase Embarazo y Lactancia). Inhibidores de CYP3A4 y CYP2D6: La administración concomitante de clorhidrato de tamsulosin con inhibidores potentes de CYP3A4 (P.Ej. ketoconazol), o en menor grado con inhibidores potentes de CYP2D6 (P.Ej. paroxetina) puede aumentar la exposición a tamsulosin (véase Interacciones). Por lo tanto, el clorhidrato de tamsulosin no se recomienda en pacientes tomando un inhibidor potente de CYP3A4 y debe usarse con precaución en pacientes tomando un inhibidor moderado de CYP3A4 (P.Ej. eritromicina), un inhibidor potente o moderado de CYP2D6, una combinación de ambos inhibidores CYP3A4 y CYP2D6, o en pacientes conocidos por ser pobres metabolizadores de CYP2D6. Insuficiencia hepática: El efecto de la insuficiencia hepática en la farmacocinética de Dutasterida no ha sido estudiada. Debido a que Dutasterida es extensamente metabolizado y tiene una vida media de tres a cinco semanas, debe tenerse precaución en la administración de DUODART a pacientes con enfermedad hepática (véase Dosis y Administración y Farmacocinética). Efectos en la capacidad de conducir y operar maquinaria: No se han realizado estudios para investigar el efecto que produce COMBODART® en la capacidad de desempeñar tareas que requiere discernimiento y habilidades motoras y cognoscitivas. Sin embargo, se les debe informar a los pacientes sobre la posible ocurrencia de síntomas relacionados con hipotensión ortostática, como mareos, cuando toman COMBODART®.
Restricciones de uso durante el embarazo y la lactancia: El uso de COMBODART® está contraindicado en mujeres. No se han realizado estudios para investigar el efecto que produce COMBODART® en el embarazo, la lactancia y la fertilidad. Las siguientes declaraciones reflejan la información disponible sobre los componentes individuales. Fertilidad: Dutasterida: En voluntarios sanos de 18 a 52 años de edad (n=27 dutasterida, n=23 placebo), se evaluaron los efectos de la administración de 0.5 mg/día de dutasterida en las características del líquido seminal, a lo largo de 52 semanas de tratamiento y 24 semanas de seguimiento posterior al tratamiento. A las 52 semanas, el promedio de reducción porcentual con respecto a la línea basal en el recuento total de espermatozoides, el volumen seminal y la motilidad de espermatozoides fue de 23%, 26% y 18%, respectivamente, en el grupo tratado con dutasterida, al realizar ajustes con respecto a los cambios observados a partir de la línea basal en el grupo placebo. La concentración y la morfología de los espermatozoides permanecieron inalteradas. Una vez transcurridas las 24 semanas de seguimiento, el promedio de cambio porcentual en el recuento total de espermatozoides de los pacientes tratados con Dutasterida siguió siendo 23% menor que el de la línea basal. Mientras que los valores promedio de todos los parámetros seminales permanecieron dentro de los intervalos normales en todo momento, por lo cual no satisficieron los criterios predefinidos de cambio clínicamente significativo (30%), dos sujetos en el grupo tratado con Dutasterida presentaron reducciones en el recuento de espermatozoides superiores al 90% del valor basal a las 52 semanas, con una recuperación parcial en el periodo de seguimiento de 24 semanas de duración. Se desconoce la significancia clínica del efecto que produce la Dutasterida en las características seminales vinculadas con la fertilidad de cada paciente. Tamsulosina: No se han evaluado los efectos del clorhidrato de Tamsulosina en el recuento o la función de los espermatozoides. Embarazo: El uso de COMBODART® está contraindicado en mujeres. Dutasterida: La Dutasterida no ha sido estudiada en mujeres porque los datos preclínicos sugieren que la supresión de los niveles circulantes de d