COPAXONE®
TEVA
Denominación genérica: Glatiramer.
Forma farmacéutica y formulación: Cada jeringa prellenada contiene: Acetato de glatiramer* 20 mg y 40 mg Vehículo, c.b.p. 1 mL. Manitol 40 mg en una solución estéril de agua para inyección. *Equivalente a 18 mg de glatiramer base.
Indicaciones terapéuticas: COPAXONE® está indicado en: El tratamiento de pacientes ambulatorios con esclerosis múltiple remitente-recurrente (EMRR): Para disminuir la frecuencia de las exacerbaciones clínicas. Para reducir el número y volumen de lesiones cerebrales activas identificadas en las imágenes por resonancia magnética (IRM).Tratamiento de los pacientes que han experimentado un primer episodio desmielinizante, acompañado por anomalías en la IRM y que se consideren en riesgo de desarrollar esclerosis múltiple clínicamente definida (EMCD), después que se excluyan diagnósticos alternativos: Para retrasar la aparición de EMCD.Para disminuir el número y volumen de lesiones cerebrales activas y la carga global de la enfermedad (como se identifica por IRM).La seguridad y eficacia de COPAXONE® en EM progresiva crónica no han sido establecidas.
Farmacocinética y farmacodinamia: Código ATC: L03AX 13. Categoría terapéutica: lnmunomodulador. DCI* o INN*: Acetato de Glatiramer. * DCI = DENOMINACIÓN COMÚN INTERNACIONAL (EL TÉRMINO EN INGLÉS ES INN = INTERNATIONAL NONPROPRIETARY NAME). Descripción: COPAXONE® es el nombre comercial del acetato de glatiramer (anteriormente conocido como copolímero-1). El acetato de glatiramer (AG), ingrediente activo de COPAXONE®, es una sal acética de polipéptidos sintéticos, que contiene una mezcla aleatorizada de cuatro aminoácidos tal y como se encuentran en la naturaleza: L-ácido glutámico, L-alanina, L-tirosina y L-lisina, con una fracción molar media de 0.129-0.153, 0.392-0.462, 0.086-0.100 y 0.300-0.374, respectivamente. El peso molecular medio del AG es 5,000-9,000 daltons y es sintetizado por polimerización de los N-carboxianhídridos de dichos aminoácidos. El AG es antigénicamente similar a la proteína básica de la mielina, un componente natural de la vaina de mielina. COPAXONE® es una solución para inyección subcutánea transparente, incolora o ligeramente amarilla, estéril y apirógena. Cada ml de solución contiene 20 mg o 40 mg de AG y 40 mg de manitol. El rango de pH de la solución es aproximadamente 5.5 a 7.0. Farmacodinamia: Los mecanismos por los que el AG ejerce sus efectos en los pacientes con esclerosis múltiple no han sido totalmente dilucidados. Sin embargo, se piensa que actúa mediante la modificación de los procesos inmunes que se cree son responsables de la patogénesis de la esclerosis múltiple. Esta hipótesis está fundamentada por los hallazgos en estudios que se han llevado a cabo para explorar la patogénesis de la encefalomielitis alérgica experimental (EAE), una condición inducida en varias especies animales mediante la inmunización contra material derivado del sistema nervioso central que contiene mielina y, a menudo, utilizado como un modelo animal experimental de esclerosis múltiple. Los estudios en animales y en sistemas in vitro sugieren que tras su administración, COPAXONE® induce la producción y activación de células T supresoras específicas de AG en la periferia.Debido a que el AG puede modificar la función inmune, existe preocupación acerca de su potencial para alterar la respuesta inmune natural. No hay evidencia de que el AG haga esto,sin embargo, este efecto no ha sido evaluado sistemáticamente.Eficacia clínica: La evidencia que apoya la eficacia de COPAXONE® en la disminución de la frecuencia de recaídas en EMRR se deriva inicialmente de 3 ensayos clínicos controlados con placebo, los cuales utilizaron una dosis de COPAXONE® de 20 mg/día. Para el síndrome clínico aislado (SCA), COPAXONE® demostró su eficacia clínica mediante un ensayo clínico controlado con placebo. La formulación de 40 mg fue probada en un estudio clínico multicéntrico, aleatorizado, doble ciego, controlado con placebo.A continuación se describen brevemente los datos más relevantes de cada uno de estos ensayos clínicos: EMRR: El estudio 1 se realizó en un solo centro. Cincuenta pacientes fueron incluidos y aleatorizados a recibir dosis diarias de COPAXONE® 20 mg por vía subcutánea o placebo (COPAXONE®: n = 25, placebo: n = 25). Los pacientes fueron diagnosticados con EMRR mediante los criterios estándar y habían tenido al menos 2 recaídas durante los 2 años inmediatamente anteriores al enrolamiento. Los pacientes eran ambulatorios, como se evidenció por una puntuación de no más de 6 en la escala expandida del estado de discapacidad de Kurtzke (EDSS), una escala estándar que va desde 0-normal a 10-muerte debido a la EM. Una puntuación de 6 se define como aquella en la que un paciente es aún ambulatorio con asistencia, una calificación de 7 significa que el paciente debe utilizar una silla de ruedas.Los pacientes fueron examinados cada 3 meses durante 2 años consecutivos, así como a lo largo de varios días cuando presentaban una supuesta recaída. Para confirmar la recaída, un neurólogo cegado al tratamiento tenía que documentar los signos neurológicos objetivos, así como documentar la existencia de otros criterios (por ejemplo, la persistencia de los signos neurológicos durante al menos 48 horas).La medida de resultado primaria especificada por el protocolo fue la proporción de pacientes en cada grupo de tratamiento que permanecieron libres de recaídas durante los 2 años del estudio; otros dos resultados fueron también especificados como criterios de valoración: la frecuencia de las recaídas durante el estudio y el cambio en el número de recaídas en comparación con el número que se produjo durante los últimos 2 años.El cuadro 1 presenta los valores de los tres resultados descritos anteriormente, así como varias medidas secundarias especificadas por el protocolo. Estos valores se basan en la población de intención para tratar (es decir, todos los pacientes que recibieron al menos 1 dosis de tratamiento y que tenían al menos una evaluación del tratamiento).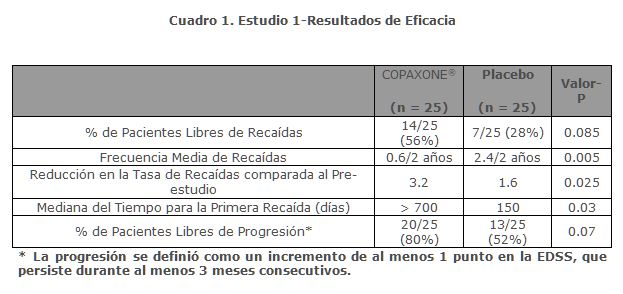
El estudio 2 fue un ensayo multicéntrico de diseño similar al primero, que se realizó en 11 centros de E.E.U.U. Un total de 251 pacientes (COPAXONE®: n = 125, placebo: n = 126) fueron incluidos. La medida de resultado primaria fue la tasa de recaída media a 2 años. El cuadro 2 muestra los valores de este resultado para la población de intención a tratar, así como varias medidas secundarias.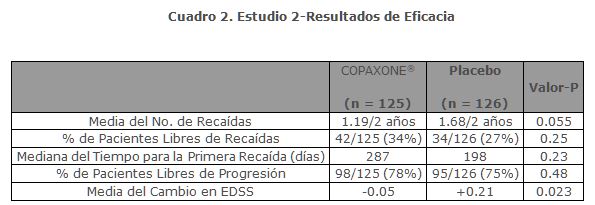
En ambos estudios, COPAXONE® exhibió un claro efecto beneficioso sobre la tasa de recaída, y es basado en esta evidencia que COPAXONE® se considera eficaz. El estudio 3 fue un estudio multinacional en el que los parámetros de resonancia magnética fueron utilizados como criterio de valoración, tanto primario como secundario. Un total de 239 pacientes con EMRR (COPAXONE®: n = 119; y el placebo: n = 120) fueron asignados al azar. Los criterios de inclusión fueron similares a aquellos en el segundo estudio, con el criterio adicional que los pacientes tenían que tener al menos una lesión realzada con gadolinio (Gd) en la RM. Los pacientes fueron tratados de una manera doble ciego durante nueve meses, durante los cuales se sometieron a una RM mensual. El criterio principal de valoración para la fase de doble ciego fue el número total acumulado de lesiones T1 realzadas con Gd a lo largo de los nueve meses. El cuadro 3 resume los resultados de la medida de valoración primaria de seguimiento durante el estudio para la cohorte de intención para tratar.
No hay evidencia actual del uso de COPAXONE® en pacientes con enfermedad primaria o secundaria progresiva. Síndrome clínico aislado: Un estudio clínico controlado con placebo (estudio PreCISe) enroló a 481 pacientes que hubieran experimentado de manera reciente (dentro de 90 días) un evento desmielinizante aislado y que tuvieran lesiones típicas de esclerosis múltiple en la RM cerebral (al menos dos lesiones cerebrales de más de 6 mm de diámetro en T2). Los pacientes fueron aleatorizados para recibir ya sea COPAXONE® 20 mg/día (n = 243) o placebo (n = 238). La medida de resultado primario fue el tiempo para el desarrollo de una segunda recaída. Los pacientes fueron seguidos durante un máximo de tres años o hasta que alcanzaron el objetivo primario. Los resultados secundarios fueron medidos por RM cerebral, incluyendo el número de nuevas lesiones en T2 y el volumen de lesión en T2. El tiempo para el desarrollo de una segunda recaída se retrasó significativamente en los pacientes tratados con COPAXONE® en comparación con placebo (cociente de riesgo = 0.55, intervalo de confianza del 95% 0.40 a 0.77; Figura 1). Las estimaciones de Kaplan-Meier del porcentaje de pacientes que desarrollaron una recaída dentro de los 36 meses fueron de 42.9% en el grupo placebo y de 24.7% en el grupo de COPAXONE®. Los pacientes tratados con COPAXONE® demostraron un menor número de nuevas lesiones en T2 en la última observación (cociente de tasas de 0.41; intervalo de confianza de 0.28 a 0.59, p < 0.0001). Además, el volumen de lesión T2 ajustado a la basal en la última observación fue menor en los pacientes tratados con COPAXONE® (razón de 0.89; intervalo de confianza de 0.84 a 0.94, p = 0.0001). Durante el periodo de más de tres años controlado con placebo, COPAXONE® retrasó la progresión del primer evento clínico de esclerosis múltiple clínicamente definida (EMCD), de acuerdo a los criterios de Poser, de una manera estadística y clínicamente significativa, lo que correspondió a una reducción del riesgo del 45% (riesgo relativo = 0.55; IC 95% [0.40-0.77], p = 0.0005). La proporción de los pacientes que desarrollaron EMCD fue de 43% en el grupo de placebo y 25% en el grupo de COPAXONE®.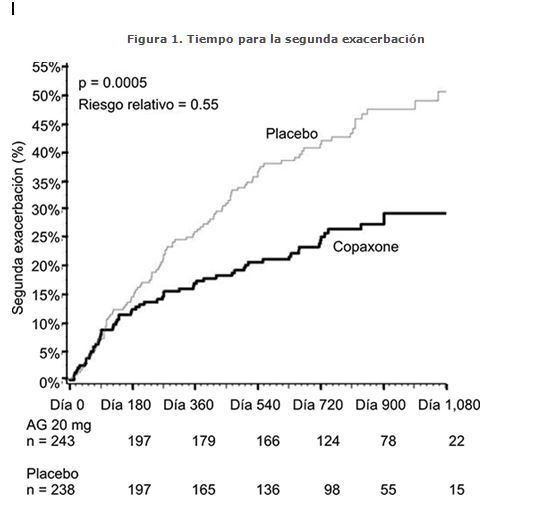
El tratamiento sólo deberá considerarse para pacientes clasificados de alto riesgo. COPAXONE® 40 mg: En un estudio clínico aleatorizado y doble ciego (estudio GALA), realizado en 142 centros en 17 países, se evalúo la seguridad y eficacia de AG 40 mg, administrado tres veces por semana (TVS), en comparación con placebo en pacientes con EMRR. Los pacientes con EMRR y al menos 1 recaída documentada los 12 meses previos a la visita de selección o con al menos 2 recaídas documentadas en los 24 meses previos a dicha visita y una puntuación en la escala EDSS ≤ 5.5 fueron asignados aleatoriamente (en una proporción 2:1) a la administración subcutánea (SC) de AG 40 mg TVS (1 mL) o placebo durante 12 meses. De los 1,524 pacientes preseleccionados, 1,404 se asignaron al tratamiento con AG 40 mg SC TVS (n = 943) o placebo (n = 461). El 93% y el 91% de los pacientes de los grupos de placebo y AG, respectivamente, completaron el estudio de 12 meses. Con AG 40 mg SC TVS se observó una reducción del 34% en el riesgo de brotes confirmados en comparación con placebo (media de la tasa anualizada de recaídas = 0.331 frente a 0.505, p < 0.0001). Los pacientes que recibieron AG 40 mg SC TVS experimentaron una reducción altamente significativa (p < 0.0001) del número acumulado de lesiones captantes de gadolinio en T1 (44.8%) y lesiones nuevas o aumentadas en T2 (34.7%) a los 6 y 12 meses. AG 40 mg SC TVS fue seguro y bien tolerado. Los eventos adversos más frecuentes en el grupo de AG fueron las reacciones en el sitio de la inyección (35.5% con AG frente al 5.0% con placebo). Los autores del estudio concluyeron que AG 40 mg SC TVS es un esquema seguro y eficaz para el tratamiento de la EMRR, con la ventaja de un menor número de inyecciones por semana. El uso de AG 40 mg SC TVS representa una opción de tratamiento para pacientes con EMRR que prefieran un esquema de inyecciones menos frecuente. Farmacocinética: La farmacocinética del AG no ha sido investigada en pacientes con EMRR o con SCA. El AG aplicado subcutáneamente se absorbe rápidamente desde el sitio de la inyección, tanto en voluntarios sanos como en animales. En animales, la absorción se produjo de una manera proporcional a la dosis y sólo el 10% de la dosis se mantuvo en el lugar de la inyección 1 hora después de la administración. A pesar de que grandes fragmentos de AG pueden ser reconocidos por anticuerpos reactivos, una gran proporción del fármaco se hidroliza a nivel local en fragmentos de pequeño peso molecular, incluyendo aminoácidos libres y oligopéptidos pequeños. Por lo tanto, se cree que las concentraciones plasmáticas son bajas en la población objetivo. Sin embargo, se han detectado niveles cuantificables de AG en el plasma de 9 de 17 voluntarios sanos que recibieron una sola dosis subcutánea supraterapéutica de 60 mg del fármaco en un estudio farmacocinético. En estos nueve pacientes, la concentración plasmática máxima (Cmáx.) fue de 69 a 605 ng/mL y el área bajo la curva concentración-tiempo del tiempo cero a seis horas (ABC10-6) fue de 1,644-67,532 ng• min/ml, el tiempo hasta la Cmáx. (Tmáx.) varió entre 15 y 30 minutos. A pesar de que las concentraciones plasmáticas de AG fueron indetectables de los 30-60 minutos después de la administración de la dosis, el fármaco se detectó de nuevo en el plasma de cuatro voluntarios en 240 a 360 minutos. Los niveles séricos fragmentos inmunoreconocibles de AG fueron < 25-50 ng/ml (es decir, por debajo del nivel de cuantificación) en las muestras de 15 de los 17 voluntarios, 24 horas después de la administración de AG. La exposición a AG parece ser similar luego de la administración de una dosis única y de una dosis múltiple (una vez al día durante un máximo de 178 días). Después de la administración de una dosis única radiomarcada de AG, la radiactividad máxima se detectó después de 1-2 horas en ratas y de 2-4 horas en monos. Los niveles radiactivos más altos fueron detectados en el estómago y la tiroides y los más bajos en el cerebro, posiblemente debido a que la alta polaridad y la naturaleza hidrofílica de AG impiden su penetración a través de la barrera hematoencefálica. Por lo tanto, el AG parece ejercer sus efectos inmunológicos en la periferia, en lugar de en el SNC (Sistema Nervioso Central). Se piensa que una proporción desconocida de AG intacto o parcialmente intacto entra en la circulación linfática, permitiendo así su distribución a los ganglios linfáticos regionales. El AG está altamente unido a proteínas plasmáticas, pero no parece desplazar o ser desplazado por la fenitoína o carbamazepina in vitro. Sin embargo, como el AG tiene el potencial de afectar la distribución de otros fármacos unidos a proteínas, se recomienda un estrecho seguimiento de los pacientes que reciben estas combinaciones de fármacos. El AG no se ha estudiado en pacientes pediátricos, ancianos o con insuficiencia renal. En animales, la excreción urinaria fue la principal vía de eliminación de la sustancia radiactiva, con heces conteniendo sólo pequeñas cantidades del fármaco. Las interacciones farmacocinéticas entre AG y otros medicamentos (incluyendo IFNs b) no han sido evaluadas formalmente. Sin embargo, en ensayos clínicos, no parece que haya interacciones perjudiciales entre AG y otros medicamentos de uso común en pacientes con EM.
Contraindicaciones: COPAXONE® está contraindicado en pacientes con hipersensibilidad conocida al AG o al manitol, embarazo y lactancia.
Precauciones generales: COPAXONE® sólo deberá administrarse subcutáneamente. COPAXONE® no debe ser administrado por vía intramuscular o intravenosa.Con el uso de COPAXONE®, se deben tener en cuenta las siguientes consideraciones y precauciones: Reacción inmediata posinyección: Aproximadamente el 16% de los pacientes expuestos a COPAXONE® en los ensayos clínicos, en comparación con el 4% de los tratados con placebo, experimentaron una serie de síntomas inmediatamente después de la inyección, que incluyó al menos dos de los siguientes: enrojecimiento, dolor torácico, palpitaciones, ansiedad, disnea, constricción de la garganta y urticaria. Los síntomas fueron generalmente transitorios y autolimitados y no requirieron tratamiento. En general, estos síntomas tienen su inicio varios meses después del inicio del tratamiento, aunque pueden ocurrir antes, y un determinado paciente puede experimentar uno o varios episodios de estos síntomas. Es incierto si cualquiera de estos síntomas represente o no realmente un síndrome específico. Durante el periodo poscomercialización, se han recibido informes de pacientes con síntomas similares que tuvieron que recibir atención médica de urgencia. Se desconoce si estos episodios son mediados por mecanismos inmunológicos o no inmunológicos, o si varios episodios similares observados en un paciente dado tienen mecanismos idénticos.El médico tratante deberá explicar al paciente que esta reacción puede ocurrir en minutos posteriores a la inyección de COPAXONE®, pudiendo presentarse cualquiera de los síntomas descritos. Asimismo, deberá explicarle que la mayoría de estos síntomas son de corta duración y se resuelven espontáneamente, sin secuelas.Si esta reacción ocurriera de modo severo, el paciente deberá interrumpir la administración de COPAXONE® y acudir con su médico o a un servicio de urgencia. Se deberá instaurar un tratamiento sintomático, el cual sería a juicio médico.Dolor de Pecho: Aproximadamente el 13% de los pacientes tratados con COPAXONE® en los ensayos clínicos, en comparación con el 6% de los pacientes tratados con placebo, experimentaron al menos un episodio de lo que fue descrito como dolor transitorio de pecho. Aunque algunos de estos episodios ocurrieron en el contexto de la reacción inmediata posinyección descrita anteriormente, muchos de ellos no ocurrieron así. La relación temporal entre este dolor y una inyección de COPAXONE® no siempre se conoce. El dolor fue transitorio (por lo general duró sólo unos minutos), a menudo no asociado con otros síntomas y no tuvo secuelas clínicas. Algunos pacientes experimentaron más de un episodio de este tipo y por lo general, dichos episodios se iniciaron por lo menos 1 mes después del inicio del tratamiento. La patogénesis de este síntoma es desconocida.Lipoatrofia y Necrosis de la Piel: Durante la experiencia poscomercialización se han reportado, en los sitios de inyección, lipoatrofia localizada y en raras ocasiones, necrosis cutáneas. La lipoatrofia puede producirse en diversos momentos después del comienzo del tratamiento (a veces después de varios meses) y se piensa que es permanente. No hay tratamiento conocido para la lipoatrofia. Para ayudar a reducir al mínimo posible estos eventos, el paciente debe seguir la técnica adecuada de inyección y rotar diariamente el sitio de aplicación de la misma.Efectos Potenciales sobre la Respuesta Inmune: COPAXONE® puede modificar la respuesta inmune, pudiendo interferir así con las funciones inmunológicas. Por ejemplo, el tratamiento con COPAXONE® puede interferir con el reconocimiento de antígenos extraños en una forma tal que comprometiera la vigilancia tumoral del cuerpo y sus defensas contra las infecciones. A la fecha, no hay evidencia de que COPAXONE® haga esto, aunque no ha habido una evaluación sistemática de este riesgo. Debido a que COPAXONE® es un material antigénico, es posible que su uso pudiera conducir a la inducción de respuestas desfavorables del huésped; a la fecha, no se ha llevado a cabo una vigilancia sistemática para estos efectos. Aunque COPAXONE® está destinado a minimizar la respuesta autoinmune a la mielina, existe la posibilidad de que la alteración continua de la inmunidad celular, debido al tratamiento crónico, pudiera tener efectos adversos.En la mayoría de los pacientes expuestos a un tratamiento diario con COPAXONE®, a la dosis recomendada, se forman anticuerpos reactivos al AG. Estos anticuerpos se detectan en suero, obteniéndose los niveles máximos después de una duración promedio del tratamiento de 3 a 4 meses; posteriormente, disminuyen sus concentraciones hasta estabilizarse en un nivel ligeramente superior al inicial. No hay evidencia que sugiera que estos anticuerpos reactivos a AG sean neutralizantes o que su formación afecte significativamente su eficacia. En un ensayo clínico controlado de 125 pacientes con EMRR aplicando 20 mg de COPAXONE® por vía subcutánea diariamente durante 2 años, los niveles séricos de lgG alcanzaron, por lo menos, 3 veces los valores basales en el 80% de los pacientes a los 3 meses de iniciado el tratamiento. A los 12 meses de tratamiento, el 30% de los pacientes todavía tenía niveles de lgG por lo menos 3 veces los valores basales, y el 90% tenía niveles por encima de la basal. Los anticuerpos fueron exclusivamente del subtipo lgG y fundamentalmente, del subtipo lgG-1. Además, estudios en ratas y monos han sugerido que se depositan complejos inmunes en los glomérulos renales. Por lo tanto, mientras estén bajo tratamiento con COPAXONE®, se deberá monitorear la función renal en aquellos pacientes que tengan algún grado de insuficiencia de este órgano.En este mismo ensayo clínico controlado con 125 pacientes, no se detectaron anticuerpos tipo lgE en ninguno de los 94 sueros probados; sin embargo, una reacción anafiláctica puede asociarse con la administración de cualquier sustancia extraña, por lo tanto, este riesgo no puede ser excluido.Otras Precauciones: Raramente se han reportado convulsiones y/o reacciones alérgicas o anafilactoides con el uso de COPAXONE®. Reacciones de hipersensibilidad severas (por ejemplo, broncospasmo, anafilaxia o urticaria) pueden ocurrir muy rara vez. Si estas reacciones son severas, deberá instaurarse un tratamiento adecuado y el tratamiento con COPAXONE® tendrá que descontinuarse.Se detectaron anticuerpos reactivos al acetato de glatiramer en el suero de los pacientes durante el tratamiento crónico diario con COPAXONE®. Los niveles máximos se alcanzaron después de una duración media de tratamientos de 3-4 meses y, a partir de entonces, disminuyó y se estabilizó en un nivel ligeramente superior a la línea de base.No hay evidencia que sugiera que estos anticuerpos reactivos al acetato de glatiramer están neutralizando o que su formación pueda afectar a la eficacia clínica de COPAXONE®.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: Clasificación en embarazo de la FDA: Categoría B que significa probablemente seguro; no hay estudios controlados en humanos y las pruebas en animales no han demostrado riesgo para el feto.Los estudios en animales son insuficientes con respecto a los efectos sobre el embarazo, desarrollo embrionario/fetal, parto o desarrollo posnatal.La administración de AG por vía subcutánea a ratas y conejas embarazadas no produjo efectos adversos sobre el desarrollo de sus crías. No hay estudios adecuados y bien controlados en mujeres embarazadas. Dado que los estudios de reproducción animal no siempre son predictivos de la respuesta humana, COPAXONE® debe utilizarse durante el embarazo sólo si es claramente necesario y queda bajo la responsabilidad del médico prescriptor. Lactancia: Los datos sobre la excreción de AG o sus metabolitos en la leche humana no están disponibles. El riesgo relativo y el beneficio para la madre y el niño deben ser tomados en consideración.
Reacciones secundarias y adversas: Ensayos clínicos controlados con placebo: En todos los ensayos clínicos, las reacciones en el sitio de la inyección fueron las reacciones adversas más frecuentes y fueron reportadas por la mayoría de los pacientes que recibieron COPAXONE®. Las reacciones en el sitio de la inyección reportadas más frecuentemente fueron eritema, dolor, induración, prurito, edema, inflamación e hipersensibilidad. Una reacción asociada con al menos uno o más de los siguientes síntomas: Vasodilatación, dolor de pecho, disnea, palpitación o taquicardia, ha sido descrita como reacción inmediata posinyección. Esta reacción puede ocurrir en minutos posteriores a la inyección de COPAXONE® (ver Precauciones generales). Todas las reacciones adversas (por lo menos un 2% más frecuentes en COPAXONE® vs. pacientes tratados con placebo) son presentadas en la tabla inferior. Estos datos se obtuvieron a partir de tres estudios clínicos pivotes, doble ciego, controlados con placebo, con un total de 269 pacientes tratados con COPAXONE® y 271 pacientes tratados con placebo durante 35 meses.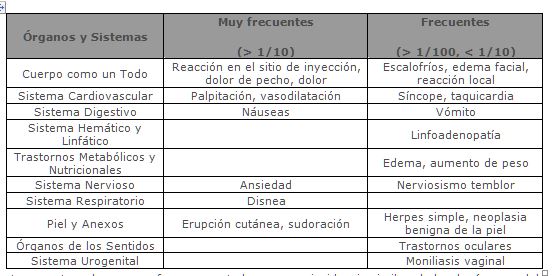
Los eventos adversos que fueron reportados con una incidencia similar al placebo (menos del 2% de diferencia) en los tres ensayos clínicos pivotes antes mencionados fueron los siguientes: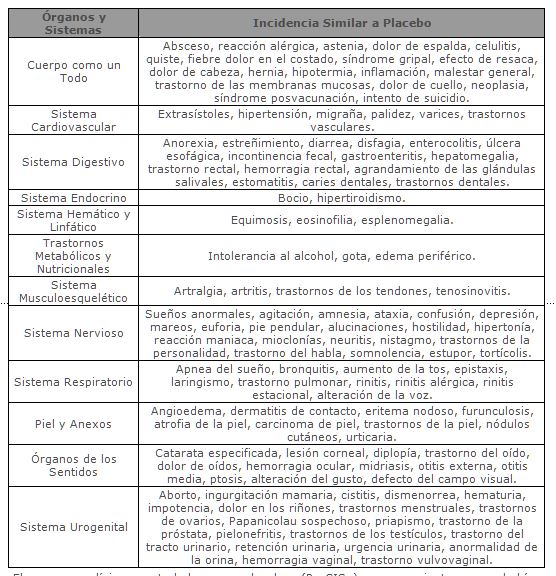
El ensayo clínico controlado con placebo (PreCISe) con pacientes que habían experimentado un primer episodio clínico (SCA) y que tenían imágenes por resonancia magnética consistentes con esclerosis múltiple incluyó 243 pacientes tratados con COPAXONE® y 238 pacientes tratados con placebo durante un máximo de 36 meses. En general, el perfil de seguridad en los pacientes con un primer episodio clínico tratados con COPAXONE® fue similar al demostrado en los tres ensayos clínicos pivotes controlados con placebo en EMRR previos. En el estudio clínico controlado con placebo (estudio GALA), los eventos adversos registrados fueron consistentes con los del perfil de seguridad conocido de la formulación de AG de 20 mg. Ensayos clínicos no controlados y experiencia poscomercialización: Notificaciones sobre reacciones adversas raras ( > 1/10000, < 1/1000) y muy raras ( < 1/10000) se obtuvieron de los pacientes con EM tratados con COPAXONE® en ensayos clínicos no controlados y con la experiencia poscomercialización de COPAXONE®. Estas notificaciones incluyen reacciones anafilácticas, convulsiones, cambios en la cantidad de glóbulos blancos y niveles elevados de enzimas hepáticas sin evidencia de secuelas clínicamente significativas. En el sitio de inyección, se han reportado lipoatrofia localizada y, en raras ocasiones, necrosis de la piel.
Interacciones medicamentosas y de otro género: Las interacciones entre COPAXONE® y otros fármacos no han sido completamente evaluadas. Los resultados de los ensayos clínicos existentes no sugieren ninguna interacción significativa de COPAXONE® con otras terapias comúnmente usadas en pacientes con EM, incluyendo el uso concomitante de corticoides durante un máximo de 28 días; sin embargo, se ha observado una mayor incidencia de reacciones en el sitio de inyección en pacientes que utilizan COPAXONE® de manera concomitante con corticosteroides. COPAXONE® no ha sido formalmente evaluada en combinación con interferón beta. Trabajos in vitro sugieren que el AG tiene gran afinidad por las proteínas plasmáticas, pero no desplaza o es desplazado por la fenitoína o la carbamazepina. Sin embargo, como COPAXONE® tiene, teóricamente, el potencial de afectar la distribución de sustancias con unión a las proteínas plasmáticas, el uso concomitante de estos fármacos debe ser monitoreado cuidadosamente.
Alteraciones en los resultados de pruebas de laboratorio: Hasta el momento, no se conoce que COPAXONE® altere o interfiera con los resultados de ninguna prueba de laboratorio.
Precauciones y relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: En un estudio de carcinogenicidad de 2 años en ratones, se les administró hasta 60 mg/kg/día de AG mediante inyección subcutánea (hasta 15 veces la dosis terapéutica humana de 20 mg/día en base a mg/m2). No se observó un aumento en las neoplasias sistémicas. En machos que recibieron dosis de 60 mg/kg/día, hubo una mayor incidencia de fibrosarcomas en los sitios de inyección. Estos sarcomas fueron asociados con daño en la piel provocado por inyecciones repetidas de un irritante sobre una zona cutánea limitada. En un estudio de carcinogenicidad de 2 años en ratas, se les administró hasta 30 mg/kg/día de AG mediante inyección subcutánea (hasta 15 veces la dosis terapéutica humana en base ena mg/m2). No se observó un aumento en los tumores.El AG no fue mutagénico en ensayos in vitro (prueba de Ames, linfoma de ratón tk). El AG fue clastogénico en dos ensayos in vitro de aberraciones cromosómicas en linfocitos humanos cultivados realizados por separado, pero no fue clastogénico en un ensayo in vivo de micronúcleos de médula ósea de ratón. No se observaron efectos adversos sobre los parámetros reproductivos o de desarrollo con la administración de AG por inyección subcutánea, antes y durante el apareamiento (machos y hembras) durante la gestación y la lactancia (hembras), a dosis de hasta 36 mg/kg/día (18 veces la dosis terapéutica humana en base a mg/m2).
Dosis y vía de administración: COPAXONE® 20 mg: La dosis recomendada en adultos es de 20 mg de AG (una jeringa precargada de COPAXONE®), administrado mediante inyección subcutánea una vez al día.COPAXONE® 40 mg: La dosis recomendada en adultos de AG de 40 mg es de una jeringa precargada de COPAXONE® (conteniendo 40 mg de AG en 1 mL de solución), administrada mediante inyección subcutánea tres veces por semana.La decisión sobre el tratamiento a largo plazo debe hacerse sobre una base individual por el médico tratante.Medidas generales a tomar en consideración: COPAXONE® sólo debe ser administrado por vía subcutánea. COPAXONE® no debe ser administrado por vía intravenosa o intramuscular.Los pacientes deben ser instruidos en las técnicas de auto-inyección y deben ser supervisados por un profesional de la salud la primera vez que se inyecte y durante los 30 minutos posteriores.Con cada aplicación se debe elegir un sitio diferente para la inyección, esto reducirá la probabilidad de cualquier irritación o dolor en el sitio de la inyección. Los sitios para la auto-inyección son el abdomen, los brazos, las caderas y los muslos.Uso en ancianos: COPAXONE® no ha sido estudiado específicamente en las personas mayores.Uso Pediátrico: No se han llevado a cabo ensayos clínicos prospectivos, aleatorizados y controlados o estudios farmacocinéticos en niños o adolescentes. Sin embargo, los limitados datos publicados sugieren que el perfil de seguridad en adolescentes de 12 a 18 años de edad que recibieron COPAXONE® es similar al observado en los adultos. El uso de COPAXONE® no está recomendado en pacientes menores de 12 años de edad, ya que no se ha establecido la seguridad y eficacia del medicamento en esta población de pacientes.Uso en Pacientes con Insuficiencia Renal: COPAXONE® no ha sido estudiado específicamente en pacientes con insuficiencia renal.Información para los Pacientes: Si el paciente olvida aplicarse una dosis de COPAXONE® a la hora que le corresponde o no puede aplicársela porque no tiene el medicamento disponible en ese momento, se la deberá aplicar lo más pronto posible en cuanto lo recuerde o en cuanto tenga a la disposición el medicamento; a partir de ese momento, la siguiente dosis se tendrá que aplicar 24 horas después para COPAXONE® 20 mg o bien, mínimo 48 horas después para COPAXONE 40 mg. Nunca se deberá administrar una dosis doble para reponer o compensar la dosis olvidada.Vía de administración: Subcutánea.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: Pocos casos de sobredosis con COPAXONE® (hasta 80 mg de AG) han sido reportados. Estos casos no se han asociado con ninguna reacción adversa distinta a las reportadas en la sección de reacciones adversas.No se han reportado casos de sobredosis con más de 80 mg de AG.En caso de sobredosis, los pacientes deben ser monitoreados y se deberán instituir terapias sintomáticas y de soporte vital adecuadas.
Presentaciones: Caja con 28 jeringas prellenadas conteniendo cada jeringa 20 mg de AG en un volumen de inyección de 1.0 ml. Caja con 12 jeringas prellenadas conteniendo cada jeringa 40 mg de AG en un volumen de inyección de 1.0 ml.
Recomendaciones sobre almacenamiento: COPAXONE® se debe almacenar en un refrigerador (2 a 8°C).Si las jeringas prellenadas no se pueden almacenar en refrigeración en las condiciones de temperatura recomendadas, se pueden entonces mantener almacenadas a temperatura ambiente (15 a 30°C) durante un periodo máximo de tiempo de hasta un mes, sin que esto tenga un impacto negativo sobre el medicamento.COPAXONE® no debe congelarse. Si una jeringa de COPAXONE® se congela, debe ser desechada.No exponga este medicamento a temperaturas elevadas. Este medicamento es sensible a la luz, por lo tanto, debe mantener las jeringas dentro de su caja. COPAXONE® no contiene conservantes. No lo use si la solución está turbia o contiene partículas en suspensión.
Leyendas de protección: No se deje al alcance de los niños. Su venta requiere receta médica. No debe administrarse en menores de 12 años. Literatura exclusiva para médicos. Reporte las sospechas de reacción adversa a los correos: farmacovigilancia@cofepris.gob.mx y safety.mexico@tevamexico.com
Nombre y domicilio del laboratorio: Hecho en Israel por: TEVA Pharmaceutical Industries Ltd 18, Eli Hurvitz Street, Industrial Zone Kfar Sava 44102, Israel Para: LEMERY, S.A. de C.V. Mártires de Río Blanco No. 54 Col. Huichapan, C.P. 16030 Deleg. Xochimilco, CDMX, México
Número de registro del medicamento: 380M2004, SSA IV