COPEGUS®
ROCHE
Denominación genérica: Ribavirina.
Forma farmacéutica y formulación: Comprimidos. Cada comprimido contiene: ribavirina 200 mg. Excipiente cbp 1 comprimido.
Indicaciones terapéuticas: COPEGUS® está indicado, en asociación con peginterferón alfa-2a (40 KD), para el tratamiento de la hepatitis C crónica en pacientes adultos, con presencia de ARN del VHC, incluyendo a los pacientes con cirrosis compensada. Para más información, consúltese la información para prescribir autorizada con peginterferón alfa-2a (40 KD).
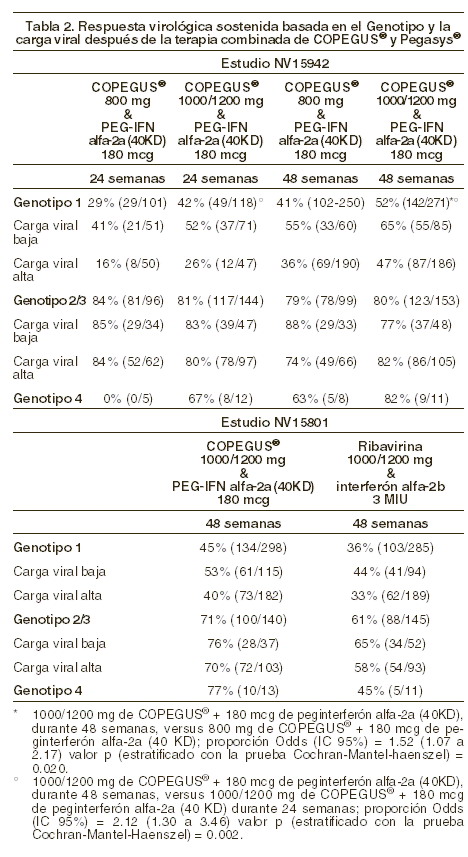
Farmacocinética y farmacodinamia: Farmacodinamia: la ribavirina es un análogo nucleosídico sintético, que presenta actividad in vitro contra ciertos virus ARN y ADN. No se conoce el mecanismo por el cual la ribavirina en asociación con peginterferón alfa-2a (40 KD) actúa contra el virus de la hepatitis C (VHC). En varios ensayos clínicos se han estudiado formulaciones orales de ribavirina como monoterapia contra la hepatitis crónica C (HCC). Los resultados de estos estudios pusieron de manifiesto que la ribavirina como monoterapia no tenía ningún efecto sobre la eliminación del virus (ARN del VHC) ni mejoraba los datos histológicos de la hepatopatía a los 6 meses de seguimiento después de 6 o 12 meses de tratamiento. Eficacia y seguridad: COPEGUS® en combinación con peginterferón alfa-2a (40 KD): hepatitis C crónica: Resultados de los estudios: la eficacia y seguridad de la combinación de COPEGUS® y peginterferón alfa-2a (40 KD) fueron demostradas en dos estudios clínicos-angulares (NV15801 + NV15942), que incluyeron un total de 2.405 pacientes. La población de los estudios incluyó pacientes con HCC confirmada con niveles detectables en sangre del RNA del virus, que no habían sido tratados previamente con interferón, con niveles elevados de transaminasas y con una biopsia de hígado consistente con hepatitis crónica por virus C. El estudio NV15801 (con 1.121 pacientes tratados) comparó la eficacia de 48 semanas de tratamiento con peginterferón alfa-2a (40 KD) (180 mg una vez por semana) y COPEGUS® (1.000/1 200 mg diarios) contra peginterferón alfa-2a (40 KD) en monoterapia o con la terapia combinada de interferón alfa-2a y ribavirina. La combinación de peginterferón alfa-2a (40 KD) y COPEGUS® fue significativamente más eficaz que la combinación de interferón alfa-2a y ribavirina o peginterferón alfa-2a (40 KD) en monoterapia (ver tabla 1). El estudio NV15942 (que incluyó 1.284 pacientes tratados) comparó la eficacia de dos períodos de tratamiento (24 y 48 semanas) y dos dosis de COPEGUS® (800 mg/día contra 1.000/1.200 mg/día). En pacientes infectados con genotipo 1, la respuesta virológica sostenida fue mayor después de 48 semanas de tratamiento que después de 24 semanas (p = 0,001) y con dosis más altas de COPEGUS® (p = 0,005). Sin embargo, para los pacientes infectados con genotipos 2 y 3, no se encontró diferencia significativa entre ambos grupos, ya sea con 48 o 24 semanas y con dosis mayores y menores de COPEGUS® (ver tabla 2). Estos patrones de respuesta no se ven influenciados por la carga viral o la presencia o ausencia de cirrosis; por consiguiente, las recomendaciones en el tratamiento son independientes de estas características basales. La respuesta virológica fue definida como niveles indetectables de RNA del virus de HCC determinado por la prueba de Cobas Amplicor™ HCV, en versión 2.0 (límite de detección de 100 copias/ml que es equivalente a 50 UI/ml) después de 6 meses de haber concluido el periodo de tratamiento.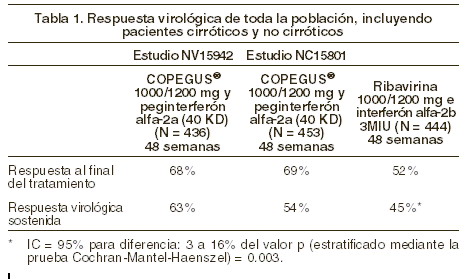
La posibilidad de considerar un acortamiento del tratamiento a 24 semanas en los pacientes con genotipos 1 y 4 se examinó a partir de una respuesta virológica rápida y sostenida observada en pacientes con respuesta virológica rápida en la semana 4 de los estudios NV15492 (ver tabla 3).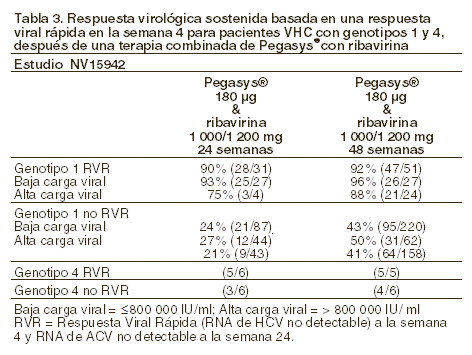
HIV-HCV coinfección: en el estudio NR15961, se distribuyeron al azar 860 pacientes co-infectados de VIH-VHC para tratarlos con 180 mg/semana de peginterferón alfa-2a y con placebo, con 180 mg/semana de peginterferón alfa-2a y 800 mg/día de ribavirina, o con 3 MIU de interferón alfa-2a, tres veces por semana, y 800 mg/día de ribavirina durante 48 semanas, seguido por un período de observación sin medicación de 24 semanas. Las respuestas virológicas sostenidas de los tres grupos de tratamiento se resumen para todos los pacientes y por genotipo en la tabla 4.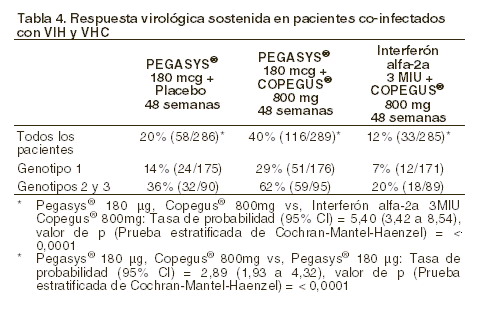
Ribavirina en combinación con interferón alfa-2a: la eficacia terapéutica del interferón alfa-2a, como monoterapia o en combinación con ribavirina por vía oral, fue comparado en estudios clínicos en pacientes que no habían sido tratados previamente y que tuvieron recaída, quienes tenían hepatitis crónica C virológica, bioquímica e histológicamente documentada. Seis meses después de finalizado el tratamiento, se observó una respuesta bioquímica y virológica sostenida, así como mejoramiento histológico. Un incremento estadísticamente significativo de 10 (de 4% a 43%; p < 0,01) en la respuesta virológica y bioquímica sostenida se observó en pacientes con recaída (M23136; N = 99). Este favorable perfil de la terapia combinada se refleja además en las velocidades de respuesta relativas al genotipo del HCC o de la carga viral basal. En los brazos de combinación y en el de interferón en monoterapia, respectivamente, la velocidad de respuesta sostenida en pacientes con HCC genotipo 1, fue de 28% versus 0% y con genotipo no 1 fue de 58% versus 8%. Adicionalmente, la mejoría histológica se favoreció por la terapia combinada. Los resultados de soporte favorables (monoterapia versus combinación; 6% vs. 48%, p < 0,04) de un pequeño estudio publicado en pacientes que no habían recibido tratamiento previo (N = 40) indica que se utilice interferón alfa-2a (3 MIU 3 veces a la semana) con ribavirina. Farmacocinética: absorción: la ribavirina se absorbe rápidamente tras la administración oral de una dosis única de COPEGUS® (Tmáx medio = 1-2 horas). La fase terminal promedio de la vida media de la ribavirina después de una dosis única de COPEGUS® oscila en el rango de 140 a 160 horas. Los datos de la literatura de la ribavirina demuestran que la absorción es extensa en un 10%, aproximadamente, de la dosis radiomarcada que se excreta por las heces. Sin embargo, la biodisponibilidad absoluta es de aproximadamente del 45 al 65%, la cual aparentemente se debe al metabolismo del primer paso. Existe una relación lineal entre la dosis y el ABCtf seguida de dosis de 200-1.200 mg de ribavirina. La depuración renal promedio después de una dosis de 600 mg de COPEGUS® por vía oral, oscila en el rango de 22 a 29 litros/hora. El volumen de distribución es de aproximadamente 4.500 litros, tras la administración de COPEGUS®. La ribavirina no se une a proteínas plasmáticas. Efecto de los alimentos: la biodisponibilidad de una sola dosis oral de 600 mg de COPEGUS® se incrementó al coadministrarla con una comida alta en grasas. Los parámetros de exposición de AUC (0-192 h) y Cmáx de la ribavirina aumentaron en 42% y 66% respectivamente si se tomaba el COPEGUS® con un desayuno alto en grasas, comparándolo con tomarlo en estado de ayuno. La relevancia clínica de estos resultados a partir de una dosis única, es desconocida. La exposición a la ribavirina después de la toma de dosis múltiples acompañadas por comida fue comparable en pacientes que recibían peginterferón alfa-2a, y COPEGUS® e interferón alfa-2a y ribavirina. Para obtener las concentraciones óptimas de ribavirina en plasma, se recomienda tomar la ribavirina con alimentos. Distribución: la ribavirina ha mostrado producir una alta variabilidad farmacocinética inter e intrapersona tras una dosis oral única de COPEGUS® (la variabilidad intrapersona de ≤25%, tanto para el ABC como para Cmáx), la cual puede deberse al metabolismo del primer paso y la transferencia hacia y dentro del compartimiento sanguíneo. El transporte de la ribavirina a través de los compartimientos no plasmáticos, ha sido estudiado extensamente sobre todo en los glóbulos rojos, y se ha identificado que la vía principal es un transportador nucleosídico de equilibrio tipo es. Este tipo de transportador se encuentra presente en todos los tipos celulares y puede contar para el gran volumen de distribución de la ribavirina. El índice de toda la sangre: la concentración plasmática de ribavirina es de aproximadamente 60 litros; el exceso de ribavirina en toda la sangre existe como nucléotidos de ribavirina secuestrados en los eritrocitos. Metabolismo: la ribavirina tiene dos vías de metabolismo: 1) una vía de fosforilación reversible, y 2) una vía de degradación que involucra una deribosilación y una hidrólisis de amida para generar como metabolito al ácido triazol-carboxílico. La ribavirina y sus dos metabolitos triazol-carboxamida y ácido triazol-carboxílico se excretan por vía renal. Tras dosis múltiples, la ribavirina se acumula extensamente en el plasma con una ABC12h seis veces mayor que la generada por dosis únicas, en base a los datos reportados en la literatura. Tras dosis orales de 600 mg dos veces al día, se alcanza el estado estacionario en aproximadamente 4 semanas, con una concentración plasmática promedio en el estado estacionario de próximamente 2.200 ng/ml. Eliminación: tras la suspensión de la dosificación, la vida media fue de aproximadamente 300 horas, lo que probablemente refleja la lenta eliminación de los compartimientos no plasmáticos. Farmacocinética en poblaciones especiales: pacientes con insuficiencia renal: la farmacocinética de dosis únicas de ribavirina se altera (aumento de ABCtf y Cmáx) en pacientes con disfunción renal, en comparación con los sujetos control, en los que la depuración renal de creatinina es superior a 90 ml/min. La depuración renal de la ribavirina administrada por vía oral disminuye sustancialmente en los pacientes con valores de > 2 mg/dl de creatinina sérica o depuración < 50 ml/min de creatinina. No existen datos suficientes acerca de la seguridad y eficacia de la ribavirina en este tipo de pacientes, sin embargo, COPEGUS® debe ser administrado con precaución e incluir medidas correctivas que incluyan suspensión del tratamiento si se presentan eventos adversos (ver Reacciones secundaria y adversas). La hemodiálisis no influye en las concentraciones plasmáticas de la ribavirina. Pacientes con insuficiencia hepática: en pacientes con disfunción hepática leve, moderada o grave, la farmacocinética de la ribavirina en dosis únicas es similar a la observada en los sujetos control sanos. Pacientes ancianos (≥ 65 años): no se han realizado estudios específicos de farmacocinética en personas ancianas. Sin embargo, en un estudio poblacional de farmacocinética, la edad no constituyó un factor clave de la cinética de la ribavirina; el factor determinante es la función renal. Pacientes menores de 18 años: no se ha realizado una evaluación exhaustiva por medio de estudios específicos de farmacocinética en pacientes menores de 18 años. COPEGUS® en asociación con peginterferón alfa-2a (40 KD) o interferón alfa-2a está indicado para el tratamiento de la HCC únicamente en pacientes de 18 o más años de edad. Raza: un estudio farmacocinético en 42 sujetos ha demostrado que no hay una diferencia clínicamente significativa en la farmacocinética de la ribavirina entre sujetos afroamericanos (n=14), hispanos (n=13) y caucásicos (n=15).
Contraindicaciones: COPEGUS® está contraindicado en pacientes con alergia a la ribavirina o a cualquiera de los excipientes. COPEGUS® no debe ser administrado ni a mujeres embarazadas ni a sus parejas. COPEGUS® está contraindicado en pacientes con hemoglobinopatías (ej. talasemia, anemia faliciforme). La terapia combinada de Pegasys® y COPEGUS® está contraindicada en pacientes con descompensación hepática. Está contraindicado el inicio de tratamiento en pacientes VIH-VHC con cirrosis, y con una calificación Child-Pugh ≥6 (ver CDS de Pegasys® para la valoración Child-Pugh). Para más información, consúltese la información para prescribir autorizada de peginterferón alfa-2a (40 KD).
Precauciones generales: De acuerdo con los resultados de los estudios clínicos, la ribavirina como monoterapia no es eficaz, por lo que COPEGUS® no debe administrarse solo. COPEGUS® en terapia combinada debe administrarse bajo la supervisión de un médico especialista. Es posible que provoque reacciones adversas que obliguen a reducir la dosis, suspender temporalmente el tratamiento o eliminarlo de forma definitiva. Riesgo teratogénico: antes de iniciar el tratamiento con COPEGUS®, el médico debe informar al paciente del riesgo teratogénico de la ribavirina, y debe hacer conciencia de la necesidad del uso de un método anticonceptivo efectivo y continuo. Además debe informar de la posibilidad de que el método anticonceptivo falle y de las consecuencias que pudieran ocurrir en el embarazo debido al tratamiento con la ribavirina. Hipersensibilidad aguda: si se presenta una reacción alérgica aguda (por ejemplo: urticaria, angioedema, broncoconstricción, anafilaxis), COPEGUS® debe ser suspendido inmediatamente e instaurar las medidas terapéuticas adecuadas. La aparición de exantemas transitorios no requiere la interrupción del tratamiento. Hemólisis y sistema cardiovascular: si existe algún tipo de deterioro o niveles anormales de la concentración de hemoglobina en la sangre, el tratamiento con COPEGUS® debe ser suspendido (ver Instrucciones especiales de dosificación). Aunque la ribavirina no tiene un efecto cardiovascular directo, la anemia asociada con COPEGUS® puede causar un deterioro de la función cardíaca o la exacerbación de los síntomas de enfermedad coronaria o ambos, por lo que COPEGUS® debe administrarse con especial precaución a los pacientes con enfermedad cardíaca preexistente clínicamente importante o inestable. El estado cardíaco del paciente debe ser examinado antes de iniciar el tratamiento y monitorear clínicamente su evolución durante éste. Si el estado cardiovascular empeora, se suspenderá la administración de ribavirina (ver Dosis y vía de administración). Se recomienda realizar un electrocardiograma a los pacientes cardiópatas antes de comenzar y durante el curso del tratamiento con COPEGUS®. Función hepática: en pacientes en los que hay evidencia de desarrollo de descompensación hepática durante el tratamiento, COPEGUS® en combinación con peginterferón alfa-2a (40 KD) debe ser suspendido. Insuficiencia renal: la terapia con COPEGUS® no debe ser iniciada en los pacientes que tengan insuficiencia renal, se encuentren o no en hemodiálisis, o continuar si el daño renal ocurre durante el tratamiento, a menos que este se considere esencial. Es recomendable que la función renal sea evaluada en todos los pacientes, previo al inicio del tratamiento, preferentemente mediante la estimación de la depuración de creatinina del paciente. Se han observado incrementos sustanciales de la concentración de ribavirina en el plasma, a las dosis recomendadas, en pacientes con valores > 2 mg/dl de creatinina sérica o con una depuración de creatinina de < 50 ml/min. No existen datos suficientes sobre la seguridad y eficacia de COPEGUS® para este grupo de pacientes que sustenten recomendaciones para la reducción de dosis, por lo que COPEGUS® debe administrarse con extrema cautela (ver Instrucciones especiales de dosificación y Farmacocinética en poblaciones especiales). Pruebas de laboratorio: antes de iniciar la terapia con COPEGUS®, a todos los pacientes se les deben realizar las pruebas estándar de hematología y bioquímica de la sangre (determinación del hemograma completo y diferencial, recuento plaquetario, electrólitos, creatinina sérica, función hepática, ácido úrico). Después de comenzada la administración de COPEGUS®, las pruebas de laboratorio deben repetirse en las semanas 2a y 4a de tratamiento y periódicamente como actividad clínica apropiada. Los siguientes valores basales deben considerarse como una guía, previa a la iniciación con COPEGUS® en combinación con peginterferón alfa-2a (40 KD): hemoglobina: ≥12 g/dl (mujeres); 13 g/dl (hombres). Plaquetas: ≥90.000/mm3. Cuenta de neutrófilos: ≥1.500/mm3. En pacientes coinfectados de VIH-VHC: CD4+ ≥ 200/ml o CD4+ ≥100 ml - > 200 ml y usando la prueba Monitor v 1.5 con Amplicor VIH-1 VIH -1 RNA < 5.000 copias/ml. Para mujeres en edad fértil: los pacientes de sexo femenino deben realizarse un examen de embarazo rutinario una vez por mes durante el tratamiento y durante los 6 meses posteriores a la finalización del mismo. Las parejas de los pacientes de sexo masculino también deben realizarse este examen de rutina con la misma periodicidad. Para más información, consúltese la información para prescribir autorizada de peginterferón alfa-2a (40 KD). Efectos sobre la capacidad para conducir vehículos y utilizar maquinaria: los efectos de COPEGUS® a este respecto son desdeñables o incluso nulos; ahora bien, el peginterferón alfa-2a (40 KD), utilizado en la terapia combinada, sí pueden tener algún efecto. Por ello, a los pacientes que presenten fatiga, somnolencia o confusión durante el tratamiento se les debe advertir que eviten la conducción de vehículos o el uso de máquinas.
Restricciones de uso durante el embarazo y la lactancia: Ni las mujeres embarazadas ni sus parejas deben tomar COPEGUS®. Los estudios con animales pusieron de manifiesto la toxicidad en la reproducción. En todas las especies animales en las que se han realizado estudios adecuados de toxicología, se ha demostrado un potencial teratógeno y embriocida significativo de la ribavirina en dosis netamente por debajo de la recomendada para el ser humano. Se observaron malformaciones del cráneo, el paladar, los ojos, la mandíbula, las extremidades, el esqueleto y el tubo digestivo. La incidencia y la gravedad de los efectos teratógenos crecían a medida que aumentaba la dosis de ribavirina. La supervivencia de los fetos y las crías disminuía. Es necesario adoptar un cuidado extremo para evitar el embarazo de las pacientes. No se empezará la administración de COPEGUS® hasta haberse obtenido la notificación de una prueba de embarazo negativa inmediatamente antes de iniciarse el tratamiento. Cualquier método anticonceptivo puede fallar. Por ello es absolutamente importante que las mujeres con capacidad para procrear y sus parejas utilicen 2 métodos anticonceptivos eficaces simultáneamente durante todo el tratamiento y los 6 meses siguientes a su conclusión; durante este período, han de efectuarse pruebas de embarazo cada mes. Si la paciente queda embarazada durante el tratamiento o el semestre siguiente después de concluido, se le informará y aconsejará acerca del riesgo importante de efectos teratógenos de la ribavirina para el feto. Pacientes del sexo masculino y sus parejas: es necesario adoptar un cuidado extremo para evitar el embarazo de las parejas de los pacientes tratados con COPEGUS®. La ribavirina se acumula en el interior celular y su eliminación fuera del cuerpo es muy lenta. En los estudios animales, la ribavirina produjo cambios espermáticos en dosis inferiores a la dosis clínica. No se sabe si la ribavirina contenida en el esperma ejerce sus conocidos efectos teratógenos tras la fertilización del óvulo; por consiguiente, se requerirá a los varones para que utilicen un preservativo (condón) a fin de reducir al mínimo la transferencia de ribavirina a sus parejas. A los pacientes de sexo masculino y a sus parejas en edad de procrear, se les aconsejará que utilicen 2 métodos anticonceptivos eficaces durante el tratamiento con COPEGUS® y los 6 primeros meses tras su terminación. Las cónyuges de los pacientes masculinos deberán tener una prueba de embarazo negativa inmediatamente antes de iniciarse el tratamiento. Se desconoce si la ribavirina se excreta en la leche materna. Debido al potencial de las reacciones adversas en lactantes, se debe tomar la decisión de discontinuar la lactancia o no iniciar la terapia.
Reacciones secundarias y adversas: El tipo y la frecuencia de los efectos adversos registrados con la terapia combinada concuerdan con el perfil toxicológico conocido del interferón alfa-2a o peginterferón alfa-2a (40 KD) y las reacciones adversas asociadas con la ribavirina. Hepatitis C crónica: la reducción de la duración del tratamiento a 24 semanas y la dosis de COPEGUS® a 800 mg resultó en la reducción de los eventos adversos serios (11% frente 3%), los retiros prematuros por razones de seguridad (13% frente 5%), y la necesidad de la modificación de la dosis de COPEGUS® (39% frente 19%), en comparación con el tratamiento de 48 semanas con COPEGUS® 1.000/1.200 mg y peginterferón alfa-2a (40 KD) 180 mg. Coinfección con VIH-VHC: en pacientes coinfectados de VIH-VHC, los eventos clínicos adversos reportados sobre el peginterferón alfa-2a, solo o en combinación con COPEGUS®, fueron similares a los observados en pacientes monoinfectados con VHC. Hay disponible información limitada sobre seguridad (N=51) en pacientes coinfectados con cuentas absolutas de CD4 + > 200/ ml. En el estudio NR 15961, las bajas causadas por eventos clínicos adversos, anormalidades de laboratorio o eventos definidores de SIDA, fueron 16% para la monoterapia con peginterferón, y de 15% para los pacientes en interferón alfa-2a en combinación con COPEGUS® 800 mg diarios, administrado durante 48 semanas. Respectivamente, 4% o 3% de los pacientes requirieron la discontinuación del tratamiento con peginterferón alfa-2a o peginterferón alfa-2a/COPEGUS® por anormalidades de laboratorio. En terapias de combinación, las modificaciones de dosis de peginterferón alfa-2a ocurrieron en 39% de los casos, y la modificación de la dosis de COPEGUS® ocurrió en 37% de los pacientes coinfectados. En 21% de los casos que recibían monoterapia con peginterferón alfa 2a y en 17% de los casos en monoterapia de peginterferón alfa 2a, se reportaron eventos adversos serios. Los tratamientos que contienen peginerferón alfa-2a han sido asociados con reducciones de la cuenta absoluta de células CD4+, sin una reducción del porcentaje de las mismas. Los índices de las células CD4+ volvieron al punto de partida durante el período de seguimiento del estudio. Los tratamientos con contenido de peginterferón alfa-2a no tuvieron un impacto negativo aparente en el control de la viremia de VIH durante el período de seguimiento. La tabla 5 muestra los efectos indeseables que ocurren con frecuencia ≥10% de los pacientes de VHC, así como en pacientes coinfectados de VIH-VHC, que habían recibido diferentes regímenes de COPEGUS® en combinación con peginterferón alfa-2a.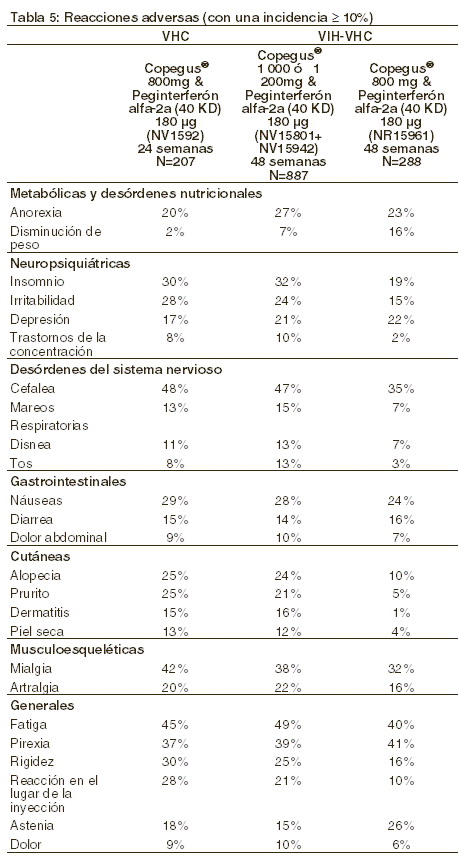
Reacciones secundarias reportadas en ≥1% pero < 10% en la combinación Pegasys®/ COPEGUS® o la monoterapia con Pegasys® fueron: infecciones e infestaciones: herpes simple, infecciones en vías aéreas superiores, bronquitis y candidiasis oral. Desórdenes en sangre y en el sistema linfático: linfadenopatía, anemia y trombocitopenia. Desórdenes endocrinos: hipotiroidismo e hipertiroidismo. Neuropsiquiátricos: alteraciones de la memoria, alteraciones en el gusto, parestesia, hipoestesia, temblor, debilidad, desórdenes emocionales, alteraciones en el humor, nerviosismo, agresión, decremento del libido, migraña, somnolencia, hiperestesia, pesadillas, síncope y ansiedad. Desórdenes en los ojos: visión borrosa, xeroftalmia, inflamación de ojos y dolor de ojos. Dolor y desórdenes en el aparato vestibular: vértigo y dolor de oído. Desórdenes cardíacos: palpitaciones, edema periférico y taquicardia. Desórdenes vasculares: bochorno. Desórdenes respiratorios, toráxicos y mediastinales: dolor de garganta, rinitis, nasofaringitis, sinusitis, disnea con esfuerzo y epistasis. Desórdenes gastrointestinales: vómito, disnea, flatulencia, boca seca, ulceración de la boca, sangrado gingival, estomatitis, disfagia e inflamación de la lengua. Desórdenes en la piel y tejido subcutáneo: desórdenes de piel, rash, eccema, soriasis, urticaria, reacción de fotosensibilidad, incremento del sudor y sudoración nocturna. Desórdenes musculoesqueléticos, del tejido conectivo y del hueso: dolor de huesos, dolor de espalda, dolor de cuello, calambres musculares, debilidad muscular, dolor musculoesquelético y artritis. Sistema reproductivo: impotencia. Desórdenes generales y condiciones en el lugar de la administración: enfermedad parecida a la influenza, malestar general, letargo, bochornos, dolor del pecho y sed. Otras reacciones adversas reportadas en ≥1% a ≤2% de pacientes con VIH-VHC que recibían la combinación Pegasys®/COPEGUS® incluyeron: hiperlactadicemia/acidosis láctica, influenza, neumonía, deterioro de los afectos, apatía, tinitus, dolor faríngolaringeo, quelitis, lipodistrofia adquirida y cromaturia. Como con otras terapias con interferón alfa, casos poco comunes o raros de los siguientes eventos adversos serios han sido reportados durante los estudios clínicos en pacientes que recibieron la combinación de Pegasys®/ COPEGUS® o monoterapia con Pegasys®: infecciones en el tracto respiratorio bajo, infecciones de la piel, otitis externa, endocarditis, suicidio, sobredosis con el medicamento, disfunción hepática, hígado graso, colangitis, neoplasia maligna hepática, úlcera péptica, sangrado gastrointestinal, reacción pancreática reversible (i.e aumento de valores de amilasa/lipasa con o sin dolor abdominal), arritmia, fibrilación auricular, pericarditis, fenómenos autoinmune (PTI, tiroiditis, soriasis, artritis reumatoide, LES), miositis, neuropatía periférica, sarcoidosis, neumonitis intersticial con desenlace fatal, embolia pulmonar, úlcera corneal, coma y hemorragia cerebral, desórdenes psicóticos y alucinaciones.
Interacciones medicamentosas y de otro género: Se han efectuado estudios de interacciones con ribavirina, en combinación con peginterferón alfa-2a (40 KD), interferón alfa-2b y antiácidos. De igual modo, las concentraciones de ribavirina son similares cuando se administra como monoterapia o en combinación con peginterferón alfa-2a (40 KD) o interferón alfa-2b. Toda posible interacción puede persistir hasta 2 meses (5 veces la vida media de la ribavirina) tras la eliminación del tratamiento con COPEGUS® a causa de su larga vida media. Los resultados de los estudios in vitro con preparaciones microsomales de hígado humano y de rata no revelaban ningún signo de metabolismo mediado por las enzimas del citocromo P450. La ribavirina no inhibe las enzimas del citocromo P450. Los estudios de toxicidad no arrojan ningún dato de inducción enzimática por la ribavirina. Por tanto, es mínimo el potencial de interacciones debidas al sistema enzimático del citocromo P450. Antiácidos: la biodisponibilidad de una dosis de 600 mg de ribavirina disminuyó tras la coadministración con antiácidos a base de magnesio, aluminio y meticona; la reducción del ABCtf fue del 14%. Es posible que la menor biodisponibilidad observada en este estudio se debiera al tránsito retardado de la ribavirina o a una variación del pH. Esta interacción no se consideró clínicamente importante. Análogos nucleosídicos: se ha demostrado in vitro que la ribavirina inhibe la fosforilación de la zidovudina y estavudina, pero se desconoce su importancia clínica. No obstante, estos resultados in vitro plantean la posibilidad de que el uso simultáneo de COPEGUS® y zidovudina o estavudina pudiera incrementar el número de VIH en la sangre. Por consiguiente, se recomienda vigilar estrechamente la concentración plasmática de ARN del VIH en los pacientes tratados a la vez con COPEGUS® y alguno de los fármacos antedichos. Si se elevan las cifras de ARN del VIH, deberá reevaluarse el uso concomitante de COPEGUS® e inhibidores de la transcriptasa inversa. En un subestudio farmacocinético de 12 semanas realizado para observar lo efectos de la ribavirina en la fosforilación de algunos inhibidores nucléosidos de la transcriptasa reversa (lamivudina, zivudina y estavudina), no se encontraron pruebas de la interacción medicamentosa observada en 47 pacientes coinfectados de VIH-VHC. La exposición del plasma a la ribavirina parece no ser afectado por la administración concomitante de inhibidores nucleósidos de la trascriptasa reversa (NRTIs). Didanosina (ddI): la ribavirina potencia el efecto antirretroviral de didanosina (ddI) in vitro y en animales mediante el incremento de la formación del anabolito activo de la trifosfatasa (ddATP). Esta observación también incrementa la posibilidad de que la administración concomitante de ribavirina y ddI pueden incrementar el riesgo de reacciones adversas relacionadas a ddI (tales como neuropatía periférica, pancreatitis y esteatosis hepática con acidosis láctica). Mientras la significancia clínica de estos hallazgos es desconocida, un estudio de ribavirina concomitante y ddI en pacientes enfermos con VIH no resultó en reducciones adicionales en la viremia o un incremento en los eventos adversos. La farmacocinética plasmática de ddI no fue significativamente afectado por la ribavirina concomitante, aunque la ddATP intracelular no fue medida. No se recomienda la administración simultánea de ribavirina y de didanosina. La exposición a la didanosina o a su metabolito activo (dideoxiadenoasina 5'-trifosfato) aumenta cuando la didanosina es coadministrada con la ribavirina. Se han reportado casos de falla hepática terminal, así como neuropatía periférica, pancreatitis y hiperlactemia sintomática/acidosis láctica a causa del uso de ribavirina.
Alteraciones en los resultados de pruebas de laboratorio: En los estudios clínicos de COPEGUS® en combinación con peginterferón alfa-2a (40 KD), la mayoría de los casos de valores analíticos alterados fueron manejados con modificaciones de la dosis (ver instrucciones especiales de dosificación). La hemólisis es el factor que define la toxicidad de la terapia con ribavirina. Un decremento en los niveles de hemoglobina a < 10 g/dl fue observado en hasta 15% de los pacientes tratados durante 48 semanas con COPEGUS® 1.000/1.200 mg en combinación con peginterferón alfa-2a (40 KD) y hasta de 19% de pacientes en combinación con interferón alfa-2a. Cuando COPEGUS® 800 mg fue combinado con peginterferón alfa-2a (40 KD) durante 24 semanas, 3% de los pacientes tuvieron un decremento en los niveles de hemoglobina hasta < 10g/dl. No se espera que los pacientes necesiten discontinuar la terapia debido sólo al decremento en los niveles de hemoglobina. En la mayoría de los casos, el decremento de la hemoglobina ocurre al inicio del período de tratamiento y se estabiliza simultáneamente con un incremento compensado en los reticulocitos. Valores de laboratorio en pacientes coinfectados de VIH-VHC: aunque casos de toxicidad hematológicas como neutropenia, trombocitopenia y anemia ocurrieron con mayor frecuencia entre pacientes de VIH-VHC, a la mayoría se le pudo controlar con modificaciones de dosis y con el uso de factores de crecimiento, así que será poco frecuente que se tenga que discontinuar el tratamiento prematuramente. La disminución de niveles ANC por debajo de 500 células/mm3 se observó en 13% y 11%, respectivamente, de los pacientes que recibieron monoterapia con peginterferón alfa-2a y terapia combinada. La disminución de plaquetas por debajo de 50.000/mm3 se observó en 10% y 8%, respectivamente, de los pacientes recibiendo monoterapia con peginterferón alfa-2a y terapia combinada. Se reportó anemia (hemoglobina por debajo de 10 g/dl) en 7% y 14%, respectivamente, de los pacientes recibiendo monoterapia con peginterferón alfa-2a y terapia combinada. Para más información, consúltese la información para prescribir autorizada de peginterferón alfa-2a (40 KD).
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Carcinogenicidad: en un estudio p53 (±) de carcinogenicidad en ratones y en otro de carcinogenicidad en ratones a 2 años con dosis máximas toleradas de 100 mg/kg/día y 60 mg/kg/día, respectivamente, se encontró que la ribavirina no es oncogénica. Basados en área de superficie corporal, esas dosis son aproximadamente 0,5 y 0,6 veces las dosis máximas de ribavirina recomendadas para humanos en 24 horas. Fertilidad: en estudios de dosis múltiples en ratones para conocer los efectos de la ribavirina en los testículos y en el esperma, se observaron anomalías espermáticas con dosis sensiblemente inferiores a las terapéuticas. Tras la suspensión de la administración, los animales se recuperaron prácticamente por completo de la toxicidad testicular en el espacio de uno o dos ciclos espermatógenos. Otros: en los estudios en animales se observó que los eritrocitos son un blanco primario de la toxicidad de la ribavirina. La anemia aparece pronto después de la administración del medicamento, pero es rápidamente reversible al cesar el tratamiento. Los estudios de genotoxicidad han demostrado que la ribavirina ejerce cierta actividad genotóxica. La ribavirina fue activa en un ensayo in vitro de transformación celular. En la prueba del micronúcleo en el ratón in vivo; se registró actividad genotóxica. La prueba letal dominante en la rata fue negativa, lo cual indicaba que, de haber mutaciones en las ratas, su transmisión no se producía a través de los gametos masculinos. No se puede excluir el riesgo de carcinogénesis en el ser humano. La administración simultánea de ribavirina y peginterferón alfa-2a (40 KD) no produjo ningún efecto tóxico inesperado en el mono. El cambio principal relacionado con el tratamiento consistió en anemia leve o moderada reversible, cuya gravedad fue mayor que la originada por uno u otro principio activo por sí solo.
Dosis y vía de administración: COPEGUS® se emplea en combinación con peginterferón alfa-2a (40 KD). La dosis exacta y la duración del tratamiento dependen del interferón empleado. Para más información, consúltese la información para prescribir autorizada de los productos con peginterferón alfa-2a (40 KD). En combinación con Pegasys® (peginterferón alfa 2a): la dosis diaria y la duración de COPEGUS® en combinación con Pegasys® debe ser personalizada basada en el genotipo viral del paciente y el peso corporal (ver tabla 6). La dosis diaria de COPEGUS® se administra oralmente en dos dosis divididas (mañana y noche) con comida. Hepatitis C crónica: la duración de la terapia combinada con ribavirina para la hepatitis C crónica depende del genotipo viral involucrado. Los pacientes infectados con VHC genotipo 1 que tengan ARN VHC detectable en la semana 4, independientemente de la carga viral pretratamiento, deberán recibir 48 semanas de terapia. Los tratamientos de 24 semanas se deben considerar para pacientes infectados con genotipo 1 y baja carga viral al inicio (LVL ≤ 800.000 IU/mL), o los que con genotipo 4 se vuelven ARN VHC negativos a la semana 4 y continúan ARN VHC negativos a la semana 24. Sin embargo, en general, un tratamiento de 24 semanas se debe considerar con un mayor riesgo de recaída que uno de 48 semanas. En estos pacientes se debe tener en cuenta la tolerancia a la terapia de combinación, así como otros factores de prognosis como el grado de fibrosis, para determinar la duración del tratamiento. Se debe considerar con más cautela aún el uso del tratamiento corto en pacientes infectados con genotipo 1 y una alta carga viral al inicio (HVL > 800.000 IU/ml) que se vuelven ARN VHC negativos en la semana 4 y que permanecen negativos en la semana 24, pues la escasa información que se tiene sugiere que estos factores impactan en forma negativa y significante, la respuesta virológica sostenida. Los pacientes infectados con VHC genotipo 2/3 deben recibir terapia de 24 semanas, sin importar la carga viral que tuvieran antes del tratamiento (ver tabla 6). La información disponible sobre pacientes con genotipos 5 o 6 es limitada; por lo tanto; se recomienda el tratamiento combinado con 1 000/1 200 mg de ribavirina durante 48 semanas.
En coinfección VIH-VHC: la dosificación recomendada para COPEGUS® en combinación con 180 mg de peginterferón alfa-2a es de 800 mg diarios durante 48 semanas, sin importar el genotipo. La seguridad y la eficacia de la terapia combinada con COPEGUS® en dosis mayores a los 800 mg o con duraciones menores a las 48 semanas no han sido estudiadas. Previsibilidad de la respuesta y la no respuesta: la respuesta virológica temprana en la semana 12, definida como un decremento de la carga viral de 2 log o niveles no detectables de ARN del VHC, se ha mostrado como un factor predictor de la respuesta sostenida (ver tabla 7).
Un valor predictivo negativo similar ha sido observado en paciente co-infectados de VIH-VHC tratados con monoterapia de peginterferón alfa-2a o en combinación con COPEGUS® (100% (130/130) o 98% (83/85), respectivamente. En pacientes coinfectados de VIH-VHC con genotipo 1 y genotipo 2/3 recibiendo terapia combinada se observaron valores predictivos positivos de 45% (50/110) y 70% (59/84) respectivamente. Instrucciones especiales de dosificación: para más información, consúltese la información para prescribir autorizada de peginterferón alfa-2a (40 KD) para ajuste de la dosis o descontinuación del tratamiento. Si hay reacciones adversas severas o anormalidades de laboratorio desarrolladas durante la terapia con COPEGUS® y peginterferón alfa-2a (40 KD), modifique la dosificación del producto hasta que las reacciones adversas se abatan. Si la intolerancia persiste después del ajuste de la dosis de COPEGUS®, la suspensión del medicamento puede ser necesaria. Para el manejo de la anemia resultante del tratamiento se desarrollaron las siguientes guías en los estudios clínicos (ver tabla 8).
Pacientes con insuficiencia renal: la farmacocinética de dosis únicas de ribavirina se altera (aumento de ABCtf y Cmáx) en pacientes con disfunción renal, en comparación con los sujetos control, en los