CUYULID
ARMSTRONG
Denominacion genérica: Ácido Alendrónico/Colecalciferol (vitamina D3).
Forma farmacéutica y formulación: Comprimidos: Alendronato sódico trihidratado equivalente a 70 mg de ácido alendrónico. Colecalciferol 140 mcg equivalente a 5600 UI de vitamina D3. Excipiente cbp 1 comprimido.
Indicaciones terapéuticas: Ácido Alendrónico/Colecalciferol está indicado para el tratamiento de la osteoporosis postmenopáusica y para ayudar a asegurar la adecuación de vitamina D. Ácido Alendrónico /Colecalciferol previene fracturas de columna y de cadera.
Farmacocinética y farmacodinamia: Farmacocinética: Aendronato: Absorción: En relación con una dosis de referencia, administrada por vía intravenosa, la biodisponibilidad oral promedio de Ácido Alendrónico/Colecalciferol en mujeres es del 0.64%, cuando se administra una dosis entre 5 y 70 mg, bajo condiciones de ayuno nocturno y dos horas antes de un desayuno estándar. La biodisponibilidad disminuye de forma similar a un estimado de 0.46% y 0.39% cuando Ácido Alendrónico/Colecalciferol se administra una hora o media hora antes de un desayuno estandarizado. En los estudios sobre osteoporosis, Ácido Alendrónico/Colecalciferol ha demostrado su eficacia cuando se administra por lo menos 30 minutos antes de la primera comida o bebida del día. El Alendronato en presentación de comprimido combinado con Ácido Alendrónico/Colecalciferol (70mg / 5,600 IU) es equivalente al comprimido formulado con 70 mg de Alendronato exclusivamente. La biodisponibilidad del Alendronato es insignificante si se administra junto con o hasta dos horas después de un desayuno estándar. La administración concomitante de Ácido Alendrónico/Colecalciferol con café o jugo de naranja reduce su biodisponibilidad aproximadamente en un 60%. En sujetos sanos, la prednisona oral (20 mg tres veces al día durante cinco días) no produce un cambio clínicamente significativo en la biodisponibilidad oral de Ácido Alendrónico/Colecalciferol (con un aumento medio del 20% al 44%). Distribución: Los estudios en ratas muestran que Ácido Alendrónico/Colecalciferol se distribuye transitoriamente por los tejidos blandos después de la administración intravenosa de 1 mg/kg de medicamento, pero luego se redistribuye rápidamente hacia el hueso o se excreta en la orina. El volumen en estado estacionario medio de distribución, sin considerar el hueso, es de al menos 28 litros en el ser humano. Las concentraciones de Ácido Alendrónico/Colecalciferol en plasma después de dosis terapéuticas orales son demasiado bajas para poder detectarlas mediante análisis ( < 5 ng / ml). La unión a proteínas en el plasma humano es aproximadamente del 78%. Biotransformación: No existe evidencia que indique que el Ácido Alendrónico/Colecalciferol sea metabolizado en animales o humanos. Eliminación: Después de una sola dosis intravenosa de Ácido Alendrónico/Colecalciferol marcada con 14C, aproximadamente el 50% de la radioactividad se excreta en la orina dentro de las primeras 72 horas y poco o nada de la radioactividad se recupera en las heces. Después de una sola dosis de 10 mg por vía intravenosa, el aclaramiento renal de Ácido Alendrónico/Colecalciferol es de 71 ml/min y el aclaramiento sistémico no excede los 200 ml/min. Las concentraciones plasmáticas disminuyen más de 95% en las seis horas después de su administración intravenosa. Se estima que la vida media terminal del medicamento en humanos pueda exceder los diez años, lo que refleja la liberación de Ácido Alendrónico/Colecalciferol desde el esqueleto. El Ácido Alendrónico no se excreta a través de los sistemas de transporte de regulación ácido - base del riñón en las ratas, por lo que no se prevé que interfiera con la excreción de otros medicamentos mediante estos sistemas en los seres humanos. Colecalciferol (vitamina D3): Absorción: En adultos sanos (varones y mujeres), después de la administración de 70 mg de Alendronato y 5,600 UI de vitamina D3 después de un ayuno nocturno y dos horas antes de una comida, el área media bajo la curva de concentración plasmática-tiempo (ABC0-80 horas) para la vitamina D3 (sin ajustar para los niveles de vitamina D3 endógena) es de 490.2 ng • h/ml. La concentración sérica máxima media (Cmáx) de vitamina D3 es de 12.2 ng / ml y el tiempo promedio hasta su concentración sérica máxima (Tmáx) es de 10.6 horas. La biodisponibilidad de 5,600 UI de vitamina D3 en la combinación de 70 mg de Alendronato/5,600 UI de vitamina D3 es similar a la administración de 5,600 UI de vitamina D3 sola. Distribución: Después de su absorción, la vitamina D3 entra en la sangre como parte de los quilomicrones. La vitamina D3 se distribuye rápidamente sobre todo al hígado donde se metaboliza a 25-hidroxivitamina D3, que es la forma principal de almacenamiento. Menores cantidades se distribuyen a los tejidos adiposo y muscular, almacenándose como vitamina D3 en estos sitios, para su posterior liberación en la circulación. La vitamina D3 en circulación se une a las proteínas de unión a vitamina D. Biotransformación: La vitamina D3 se metaboliza rápidamente por hidroxilación en el hígado a 25-hidroxivitamina D3, y posteriormente es metabolizada en el riñón a 1,25-dihidroxivitamina D3, que representa a la forma biológicamente activa de la vitamina. La hidroxilación continúa antes de la eliminación. Un pequeño porcentaje de vitamina D3 sufre glucuronidación antes de su eliminación. Eliminación: Cuando se administró vitamina D3 radioactiva a sujetos sanos, la excreción urinaria media de radioactividad después de 48 horas fue de 2.4%, y la excreción fecal media de radioactividad después de 4 días fue de 4.9%. En ambos casos, la radioactividad excretada fue casi exclusivamente en forma de metabolitos del fármaco original. La vida media de la vitamina D3 en el suero después de una dosis oral de (70 mg/2.800 UI) es de aproximadamente 24 horas. Características en los pacientes: Estudios clínicos muestran que el Alendronato que no se llega a depositar en los huesos, es rápidamente excretado a través de la orina. No se observaron datos de saturación en la captación ósea después de administrar en forma crónica, dosis intravenosas acumulativas de hasta 35 mg/kg en animales. Aunque no existe información clínica disponible, es probable que, al igual que sucede en animales de experimentación, la eliminación renal de Ácido Alendrónico/Colecalciferol se reduzca en pacientes con función renal alterada. Por lo tanto, es de esperar una acumulación algo mayor de Ácido Alendrónico/Colecalciferol en los huesos en pacientes con insuficiencia renal (ver Dosis y vía de administración). Farmacodinamia: Grupo farmacoterapéutico: Medicamentos para el tratamiento de enfermedades óseas, bifosfonatos, combinaciones. Código ATC: M05BB03. Ácido Alendrónico /Colecalciferol es un comprimido que contiene la combinación de dos ingredientes activos: Alendronato y Colecalciferol (vitamina D3). Alendronato: Es un bifosfonato que se une a la hidroxiapatita del hueso e inhibe en forma específica, la resorción ósea mediada por los osteoclastos, sin efecto directo en la formación del hueso, aunque este último proceso, disminuya finalmente debido a que la relación entre la resorción y la formación óseas van aparejadas durante el intercambio óseo. Los estudios preclínicos han demostrado la localización preferente de Alendronato en sitios de resorción activa. La actividad de los osteoclastos se inhibe, pero el reclutamiento o fijación de los osteoclastos no se ve afectada. El hueso formado durante el tratamiento con Ácido Alendrónico/Colecalciferol es de calidad normal. Colecalciferol (vitamina D3): La vitamina D3 se produce en la piel por conversión del 7-dehidrocolesterol a vitamina D3 por la luz ultravioleta. A falta de una adecuada exposición al sol, la vitamina D3 es un nutriente alimenticio esencial. La vitamina D3 se convierte a 25-hidroxivitamina D3 en el hígado, y se almacena hasta que se necesite. La conversión a la hormona activa movilizadora de calcio, la 1,25-dihidroxivitamina D3 (calcitriol) en el riñón, está altamente regulada. La principal acción de la 1,25 dihidroxivitamina D3 es aumentar la absorción intestinal tanto de calcio como de fosfato, así como regular los niveles séricos de calcio y la excreción renal de fosfato, la formación de hueso y la resorción ósea. La vitamina D3 se requiere para la formación normal del hueso. La insuficiencia de vitamina D se desarrolla cuando la exposición al sol y su ingesta a través de la dieta son insuficientes. La insuficiencia renal se asocia con un balance negativo de calcio, pérdida de hueso y un mayor riesgo de fractura ósea. En los casos graves, el déficit resulta en hiperparatiroidismo secundario, hipofosfatemia, debilidad muscular proximal y osteomalacia, aumentando aún más el riesgo de caídas y fracturas en individuos osteoporóticos. La vitamina D suplementaria reduce estos riesgos y sus consecuencias. La osteoporosis se define como una densidad mineral ósea (DMO) de la columna vertebral o de la cadera 2.5 desviaciones estándar (DE) por debajo del valor medio de una población joven normal o como una fractura previa por fragilidad osea, independientemente de la DMO. Estudios clínicos Alendronato / vitamina D3: El efecto de la dosis más baja de 70 mg de Alendronato / y 2,800 UI de vitamina D3 sobre el estado de la condición de la vitamina D se demostró en un estudio multinacional de 15 semanas, que incluyó a 682 mujeres post-menopáusicas con osteoporosis (25-hidroxivitamina D basal: promedio 56 nmol / l [22.3 ng / ml], rango 22.5 a 225 nmol / l [9-90 ng / ml]). Las pacientes recibieron la dosis más baja (70 mg/2,800 UI) (n = 350) o Alendronato 70 mg (n = 332) una vez a la semana y se les prohibieron los suplementos adicionales de vitamina D. Después de 15 semanas de tratamiento, los principales resultados observados fueron: 1. Concentración sérica promedio de 25-hidroxivitamina D fue significativamente más altos (26%) en el grupo (70 mg/2,800 UI) (56 nmol / l [23 ng / ml]), que en el grupo que solo tomó Alendronato (46 nmol / l [18.2 ng / ml]). 2. El porcentaje de pacientes con deficiencia de vitamina D (25-hidroxivitamina D sérica < 37.5 nmol / l [ < 15 ng / ml]), disminuyó significativamente en un 62.5% en el grupo (70 mg/2,800 UI), en comparación con el grupo con Alendronato solo (12% vs. 32%, respectivamente). 3. El porcentaje de pacientes con deficiencia de vitamina D (25-hidroxivitamina D sérica < 22.5 nmol / l [ < 9 ng / ml]), disminuyó significativamente en un 92% con (70 mg/2,800 UI) frente al Alendronato sólo (1% frente a 13 %, respectivamente). 4. El promedio de 25-hidroxivitamina D en pacientes con deficiencia de vitamina D al inicio del estudio (25-hidroxivitamina D, 22.5 a 37.5 nmol / l [9 a < 15 ng / ml]) aumentó de 30 nmol / l (12.1 ng / ml) a 40 nmol / l (15.9 ng / ml) en el grupo (70 mg/2,800 UI) (n = 75) y disminuyó de 30 nmol / l (12.0 ng / ml) al inicio del estudio a 26 nmol / l (10.4 ng / ml) en el grupo de Alendronato solo (n = 70) a las 15 semanas de tratamiento. 5. Sin diferencias en los niveles séricos de calcio, fósforo o calcio en orina de 24 horas entre los grupos de tratamiento. El efecto de la dosis más baja de Alendronato 70 mg / vitamina D3 2,800 UI) más un aporte adicional de 2,800 UI de vitamina D3 para un total de 5,600 UI (cantidad máxima de vitamina D3), una vez por semana, se demostró en un estudio de 24 semanas, cuya extensión incluyó a 619 mujeres postmenopáusicas con osteoporosis. Los pacientes en el grupo de vitamina D3 2,800 recibieron (70 mg/2,800 UI) (n = 299) y pacientes en el grupo de vitamina D3 5,600, recibieron (70 mg/2.800 UI) más un 2,800 UI adicional de vitamina D3 (n = 309) una vez a la semana. A las pacientes, se les permitió tomar suplementos adicionales de vitamina D después de 24 semanas de tratamiento. Los principales resultados fueron: 1 La concentración sérica promedio de 25-hidroxivitamina D fue significativamente mayor en el grupo de vitamina D3 5,600 (69 nmol / l [27.6 ng / ml]) que en el grupo de vitamina D3 2,800 (64 nmol / l [25.5 ng / ml]). 2. El porcentaje de pacientes con deficiencia de vitamina D fue de 5.4% en el grupo de vitamina D3 2,800 frente a 3.2% en el grupo de vitamina D3 5,600 a través de la extensión de 24 semanas. 3. El porcentaje de pacientes con deficiencia de vitamina D fue del 0.3% en el grupo de vitamina D3 2, 800 frente a cero en el grupo D3 5,600. 4. No hubo diferencias en los niveles séricos de calcio, fósforo o calcio en orina de 24 horas entre los grupos de tratamiento. 5. La diferencia en el porcentaje de pacientes con hipercalciuria al final de la extensión de 24 semanas no fue estadísticamente significativa entre los grupos de tratamiento. Estudios con Alendronato: La equivalencia terapéutica de Ácido Alendrónico/Colecalciferol 70 mg una vez por semana (n = 519) y Ácido Alendrónico/Colecalciferol 10 mg al día (n = 370) se demostró en un estudio multicéntrico de un año de duración en mujeres posmenopáusicas con osteoporosis. Los principales resultados fueron los siguientes: 1. El aumento promedio sobre un valor basal, de la DMO de columna lumbar al año fue del 5.1% (IC del 95%: 4.8 - 5.4%), en el grupo de 70 mg una vez a la semana y el 5.4% (IC del 95%: 5.0 5.8%) en el grupo de 10 mg diarios. 2. El aumento promedio de DMO fue del 2.3% y 2.9% en el cuello femoral y del 2.9% y 3.1% en toda la cadera en el grupo de pacientes tratadas con 70 mg una vez por semana y 10 mg diarios, respectivamente. 3. Los dos grupos de tratamiento también fueron similares con respecto a los aumentos de la DMO en otras zonas esqueléticas. Los efectos del Alendronato sobre la masa ósea y la incidencia de fracturas en mujeres post-menopáusicas se investigaron en dos estudios iniciales de eficacia de diseño idéntico (n = 994), así como en el Ensayo de Intervención en Fracturas (FIT por sus siglas en inglés: Fracture Intervention Trial) (FIT: n = 6,459). Los principales resultados fueron los siguientes: 1. En estudios iniciales sobre eficacia, el aumento promedio en la DMO con Ácido Alendrónico/Colecalciferol 10 mg/día en relación con el placebo a tres años de tratamiento fueron del 8.8%, 5.9% y 7.8% en la columna, cuello femoral y trocánter, respectivamente. 2. La DMO corporal total también aumentó significativamente. 3. Hubo una reducción del 48% en la proporción de pacientes tratados por una o más fracturas vertebrales en pacientes tratadas con Ácido Alendrónico /Colecalciferol 10 mg diarios, con respecto a las pacientes tratadas con placebo. En la extensión de dos años de estos estudios, la DMO en la columna y el trocánter continuó aumentando y la DMO en el cuello femoral, se mantuvieron todo el tiempo. El estudio FIT consistió en dos ensayos controlados con placebo utilizando Ácido Alendrónico/Colecalciferol diariamente (5 mg diarios durante dos años y 10 mg diarios durante uno o dos años adicionales): FIT 1: Un estudio de tres años de duración realizados en 2,027 pacientes que hubieran presentado al menos una fractura vertebral durante este periodo. En este estudio la ingesta diaria de Ácido Alendrónico/Colecalciferol redujo la incidencia en ≥ 1, las nuevas fracturas vertebrales en un 47% (Ácido Alendrónico/Colecalciferol 7.9% frente a placebo 15.0%). Además, se encontró una reducción estadísticamente significativa en la incidencia de fracturas de cadera (1.1% frente a 2.2%, respectivamente, una reducción de 51%). FIT 2: Un ensayo de cuatro años de duración, realizado en 4,432 pacientes con masa ósea baja pero sin fracturas vertebrales en condiciones basales. En este estudio, se observó una diferencia significativa en el análisis del subgrupo de mujeres con osteoporosis (el 37% de la población total se corresponde con la definición anterior de osteoporosis) en la incidencia de fracturas de cadera (Ácido Alendrónico/Colecalciferol 1.0% frente a placebo 2.2 %, una reducción de 56%) y en la incidencia de ≥ 1 fractura vertebral (2.9% frente a 5.8% respectivamente, una reducción de 50%). Pruebas de laboratorio: En estudios clínicos, se observaron descensos asintomáticos, leves y transitorios del calcio sérico y del fosfato en aproximadamente el 18% y 10% respectivamente, de los pacientes que tomaron Ácido Alendrónico/Colecalciferol 10 mg/día en comparación con aproximadamente el 12% y el 3% de los que tomaron placebo. Sin embargo, las incidencias de descensos de calcio sérico a < 8.0 mg/dl (2.0 mmol / l) y fosfato sérico a ≤ 2.0 mg/dl (0.65 mmol / l) fueron similares en ambos grupos de tratamiento.
Contraindicaciones: Hipersensibilidad a los principios activos o a cualquiera de los excipientes. Anormalidades esofágicas y otros factores que retrasan el vaciamiento esofágico, como estenosis o la acalasia. Incapacidad para deglutir. Incapacidad para quedarse parado o sentado durante al menos 30 minutos. Hipocalcemia (ver sección Precauciones Generales). Insuficiencia renal con aclaramiento de creatinina < 0.58 ml/s [ < 35 ml/min].
Precauciones generals: General: Para facilitar la llegada al estómago y así reducir el potencial de irritación esofágica, los pacientes deben ser instruidos para ingerir cada tableta de alendronato/colecalciferol con un vaso lleno de agua (200 a 250 ml) y no acostarse durante al menos 30 minutos y hasta después de su primera comida del día. Los pacientes no deben masticar o chupar la tableta debido a un potencial de ulceración orofaríngea. Los pacientes deben ser instruidos específicamente de no tomar alendronato/colecalciferol al acostarse o antes de levantarse por la mañana. Los pacientes deben ser informados de que el incumplimiento de estas instrucciones puede aumentar su riesgo de problemas esofágicos. Los pacientes deben ser instruidos de que si desarrollan síntomas de la enfermedad esofágica (como dificultad o dolor al tragar, dolor retroesternal o aparición o empeoramiento del ardor esofágico) deben dejar de tomar alendronato/colecalciferol inmediatamente y consultar a su médico. Se deben tomar en cuenta las causas de osteoporosis distintas a la deficiencia de estrógenos, el envejecimiento y el uso de glucocorticoides. Osteonecrosis mandibular: La osteonecrosis de la mandíbula (ONM) ha sido reportada en pacientes con cáncer que reciben regímenes de tratamiento incluyendo los bisfosfonatos. La mayoría de los reportes ocurrieron después de extracciones dentales con cicatrización retrasada e involucraron a pacientes con cáncer tratados con bisfosfonatos intravenosos. Muchos de estos pacientes estaban recibiendo también quimioterapia y corticosteroides. Sin embargo, algunos casos también ocurrieron en pacientes recibiendo tratamiento con bisfosfonatos por vía oral para la osteoporosis postmenopáusica y otros diagnósticos. La mayoría de los casos reportados han sido asociados con procedimientos dentales tales como la extracción dental. Muchos tuvieron signos de infección local, incluyendo la osteomielitis. Un examen dental con una apropiada odontología preventiva debe ser considerado antes del tratamiento con bisfosfonatos en pacientes con factores de riesgo concomitantes. Los factores de riesgo conocidos para osteonecrosis de la mandíbula incluyen un diagnóstico de cáncer, terapias concomitantes (por ejemplo, quimioterapia, radioterapia, corticosteroides, inhibidores de la angiogénesis, fármacos inmunosupresores), potencia del bifosfonato (más alta para el ácido zoledrónico), la vía de administración y la dosis acumulativa, higiene oral deficiente, trastornos comórbidos (por ejemplo, enfermedad periodontal y/u otra enfermedad dental preexistente, anemia, coagulopatía, infección, diabetes mellitus), tabaquismo, procedimientos dentales invasivos y dentaduras mal ajustadas y consumo excesivo de alcohol. Un examen dental con un adecuado tratamiento odontológico preventivo debe ser considerado antes del tratamiento con bifosfonatos orales en pacientes con estado dental deficiente. Mientras el tratamiento continúe, estos pacientes deben evitar procesos dentales invasivos, si es posible. En los pacientes que desarrollen osteonecrosis mandibular durante la terapia con bifosfonatos, la cirugía dental puede agravar la situación. Para los pacientes que requieran intervenciones dentales, no hay datos disponibles que sugieran si la suspensión del tratamiento con bifosfonatos reduce el riesgo de osteonecrosis de la mandíbula. La valoración clínica del facultativo, debe orientar sobre cómo proceder con cada paciente según la valoración beneficio/riesgo individual. Durante el tratamiento con bifosfonatos, todos los pacientes deben ser alentados a mantener una buena higiene oral, realizarse chequeos dentales rutinarios y reportar cualquier síntoma oral, tales como la movilidad dental, dolor, o hinchazón. Los pacientes que desarrollen osteonecrosis de la mandíbula deben recibir tratamiento antibiótico apropiado y/o cirugía oral, y la suspensión del tratamiento con bisfosfonatos debe considerarse con base en la evaluación individual de riesgos y beneficios. La cirugía dental puede agravar la situación. Para los pacientes que requieren procedimientos dentales (por ejemplo, extracción de dientes, implantes dentales), no hay datos definitivos disponibles para establecer si la interrupción del tratamiento con bisfosfonatos reduce el riesgo de osteonecrosis de la mandíbula. El juicio clínico del médico tratante y/o cirujano oral debe guiar el plan de manejo, incluyendo el tratamiento con bisfosfonatos, de cada paciente en función de la evaluación individual de riesgos y beneficios. La Osteonecrosis del conducto auditivo externo se ha reportado asociado principalmente al tratamiento de largo plazo con bisfosfonatos. Los posibles factores de riesgo para la osteonecrosis del conducto auditivo externo incluyen el uso de esteroides y la quimioterapia y/o factores de riesgos locales como infección o traumatismo. La posibilidad de una osteonecrosis del conducto auditivo externo debe considerarse en pacientes que reciben bisfosfonatos y que se presentan con síntomas del oído incluyendo infecciones crónicas del oído. Dolor musculoesquelético: Se ha informado de dolor en huesos, articulaciones y/o músculos en pacientes tratados con bifosfonatos. En la experiencia post-comercialización, estos síntomas raramente fueron graves y/o incapacitantes (ver reacciones secundarias). Sin embargo, estos reportes han sido poco frecuentes. Dento de esta categoría de medicamentos, se encuentra el alendronato sódico. La mayoría de los pacientes afectados fueron mujeres posmenopáusicas. El tiempo de aparición de los síntomas fue variable, desde un día hasta varios meses después de comenzar el tratamiento. La mayoría de los pacientes aliviaron el dolor después de interrumpir el tratamiento. Un subgrupo presentó recurrencia de los síntomas al exponerse nuevamente al mismo medicamento u otro bifosfonato. En estudios clínicos controlados contra placebo de alendronato sódico, los porcentajes de pacientes con estos síntomas fueron similares en los grupos de alendronato sódico y del placebo. Fracturas atípicas del fémur: Se han reportado fracturas femorales de baja energía, de localización subtrocantérica y diafisaria asociadas al tratamiento de largo plazo con bifosfonatos (el tiempo hasta la aparición en la mayoría de los reportes osciló entre 18 meses a 10 años). Algunas eran fracturas de estrés (reportadas como fracturas por insuficiencia), que ocurrieron en ausencia de trauma aparente. Las fracturas pueden ocurrir en cualquier lugar a lo largo del fémur, desde justo debajo del trocánter menor hasta justo por encima de la cresta supracondílea. Estas fracturas se producen después de un trauma mínimo o sin este, algunos pacientes experimentan dolor en el muslo o en la ingle, a menudo asociado con imágenes características que indican fractura por sobrecarga, semanas o meses antes de presentar una fractura femoral completa. Las fracturas son a menudo bilaterales, por lo que el fémur del lado opuesto debe ser examinado en los pacientes tratados con bifosfonatos que hayan sufrido una fractura de la diáfisis femoral. También se ha notificado un bajo índice de curación de estas fracturas. Durante el tratamiento con bifosfonatos, debe instarse al paciente a informar cualquier dolor de muslo, cadera o ingle, cualquier paciente que presente dichos síntomas debe ser evaluado en busca de una fractura incompleta de fémur. Los pacientes con sospecha de fracturas por estrés deben ser evaluados, incluyendo la evaluación de las causas y factores de riesgo de fracturas por estrés (por ejemplo, la deficiencia de vitamina D, malabsorción, el uso de glucocorticoides, artritis o fractura de extremidad inferior, fractura por estrés anterior, ejercicio extremo o aumentado, diabetes mellitus, abuso crónico de alcohol), y recibir una atención ortopédica apropiada. La interrupción de la terapia con alendronato en pacientes con fracturas por estrés se debe considerar con base en una evaluación individual de riesgos y beneficios. Insuficiencia Renal: No se recomienda usar Ácido Alendrónico/Colecalciferol en pacientes con insuficiencia renal con un índice de filtración glomerular (IFG) menor a 35 ml/min (ver la Dosis y vía de administración). Endocrinología y metabólico: Alendronato sódico: La hipocalcemia debe corregirse antes de iniciar el tratamiento con Ácido Alendrónico/Colecalciferol (ver contraindicaciones). Otros trastornos que afectan al metabolismo mineral (como la deficiencia de vitamina D e hipoparatiroidismo) también deben ser tratados eficazmente antes de comenzar con el uso de Ácido Alendrónico/Colecalciferol. El contenido de vitamina D en Ácido Alendrónico/Colecalciferol no es adecuado para la corrección de la deficiencia de vitamina D. En los pacientes con estas condiciones, el calcio sérico y los síntomas de hipocalcemia deben vigilarse durante el tratamiento con Ácido Alendrónico/Colecalciferol. La hipocalcemia sintomática se ha reportado en raras ocasiones, tanto en pacientes con o sin factores predisponentes conocidos. Los pacientes deben ser advertidos de que reporten a sus médicos cualquier síntoma de hipocalcemia, como parestesias o espasmos musculares. Los médicos deben evaluar cuidadosamente a los pacientes que desarrollan hipocalcemia durante el tratamiento con alendronato/colecalciferol debido a condiciones predisponentes. Debido a los efectos positivos del Alendronato sobre el aumento de la mineralización ósea, los niveles séricos de calcio y fosfato pueden disminuír especialmente en pacientes que toman glucocorticoides en quienes la absorción de calcio puede estar reducida. Estos casos son por lo general menores y asintomáticos. Sin embargo, se han notificado casos raros de hipocalcemia sintomática, que ocasionalmente han sido graves, que aparecen en pacientes con factores predisponentes (por ejemplo hipoparatiroidismo, deficiencia de vitamina D y malabsorción de calcio) (ver Reacciones secundarias). Colecalciferol: Los pacientes con malabsorción pueden asimilar inadecuadamente la vitamina D3. Alendronato/colecalciferol solo no debe ser utilizado para tratar la deficiencia de vitamina D (comúnmente definida como una concentración sérica de 25-hidroxivitamina D < 22.5 nmol/l o 9 ng/ml). Los pacientes que sufren de osteoporosis están en un mayor riesgo de insuficiencia de vitamina D, especialmente los mayores a los 70 años de edad, confinados al hogar o enfermos crónicos, pueden recibir suplementación con vitamina D, además de la que se proporciona con la administración del alendronato/colecalciferol (véase la sección Dosis y Vía de Administración). Los que viven en latitudes altas (incluyendo la mayor parte de Canadá) también pueden necesitar suplementación adicional. Los pacientes con síndrome de mala absorción intestinal pueden requerir también dosis más altas de suplementos de vitamina D y considerar la medición de la concentración de 25-hidroxivitamina D sérica. La vitamina D3 puede incrementar la magnitud de la hipercalcemia y/o hipercalciuria cuando es administrada a pacientes con alguna enfermedad asociada a una sobreproducción no regulada de calcitriol (por ejemplo leucemia, linfoma, sarcoidosis). Los niveles de calcio, séricos y en orina, deben ser monitorizados en estos pacientes. Gastrointestinal: Alendronato/colecalciferol, al igual que otros productos que contienen bisfosfonatos, puede causar irritación local de la mucosa gastrointestinal superior. Debido a que existe el riesgo potencial de empeoramiento de la enfermedad subyacente, se debe tener precaución cuando el alendronato/Colecalciferol se administra a pacientes con trastornos activos del tubo digestivo superior, como disfagia, patología esofágica, gastritis, duodenitis, úlceras o con una historia reciente (en el año anterior) de enfermedad gastrointestinal grave como úlcera péptica, hemorragia gastrointestinal activa, o cirugía del tracto gastrointestinal superior diferente de la piloroplastía (ver Contraindicaciones). En los pacientes con esófago de Barrett, los médicos prescriptores deben considerar los beneficios y riesgos potenciales de Ácido Alendrónico/Colecalciferol en función del paciente. Los eventos adversos esofágicos (algunas veces graves y que requieren hospitalización), como esofagitis, úlceras y erosiones esofágicas, raramente seguidas de estenosis, se han reportado en pacientes que reciben Ácido Alendrónico/Colecalciferol. Por lo tanto, los médicos deben estar atentos a cualquier signo o síntoma que sugiera una posible reacción esofágica y los pacientes deben ser aconsejados para dejar de consumir Ácido Alendrónico/Colecalciferol y buscar atención médica si desarrollan síntomas de irritación esofágica, como disfagia, dolor al deglutir o dolor retroesternal o la aparición o empeoramiento de pirosis (ver reacciones secundarias). El riesgo de reacciones adversas esofágicas graves parece ser mayor en los pacientes que no toman alendronato/Colecalciferol y/o que no lo ingieren junto con un vaso lleno (200 a 250 ml) de agua, y/o que siguen tomando alendronato/Colecalciferol después de desarrollar síntomas indicativos de irritación esofágica. Es muy importante que el paciente reciba y comprenda las instrucciones completas de administración (ver Dosis y vía de administración). Aunque no se observó aumento del riesgo en ensayos clínicos extensos con alendronato/Colecalciferol, se han notificado informes poco frecuentes (post-comercialización) de úlceras gástricas y duodenales, algunos de los cuales fueron graves y con complicaciones (ver reacciones secundarias). Oftalmológico: Los trastornos oculares incluyendo conjuntivitis, uveítis, epiescleritis y escleritis se han reportado con la terapia de alendronato. Los pacientes con eventos oculares distintos a la conjuntivitis no complicada, deben ser remitidos a un oftalmólogo para su evaluación. Si se observan síntomas inflamatorios oculares, podrá ser necesario suspender el tratamiento. Poblaciones especiales: Pediátricas ( < 18 años de edad): Alendronato/colecalciferol no se ha estudiado en pacientes < 18 años de edad y no debe ser administrado a ellos. Geriátricas: Alendronato sódico: En estudios clínicos no hubo diferencias relacionadas con la edad en los perfiles de eficacia o seguridad de alendronato sódico. Colecalciferol: Las necesidades diarias de vitamina D3 podrán incrementarse en las personas de edad avanzada. Monitoreo y pruebas de laboratorio: No aplica. Efectos en la capacidad de conducir y operar maquinaria: No hay estudios sobre los efectos en la capacidad para conducir y utilizar máquinas. Sin embargo, ciertas reacciones adversas que se han reportado con alendronato/colecalciferol (por ejemplo, mareos, vértigo, trastornos visuales y dolor severo de huesos y articulaciones) pueden afectar a la capacidad de conducir u operar maquinaria de algunos pacientes. Las respuestas individuales a alendronato/colecalciferol pueden variar.
Restricciones de uso durante el embarazo y la lactancia: Alendronato/Colecalciferol está indicada solamente para mujeres postmenopáusicas, por lo que no debe ser usado en el embarazo o en el periodo de lactancia. Uso en el embarazo: Alendronato/colecalciferol no ha sido estudiado en mujeres embarazadas y no debe administrarse a ellas. Los estudios en animales no muestran efectos dañinos directos sobre el embarazo, desarrollo embrionario/fetal o desarrollo postnatal. El alendronato/Colecalciferol causó distocia relacionada a hipocalcemia en una prueba realizada en ratas preñadas. Estudios en animales han mostrado hipercalcemia y toxicidad reproductiva con dosis altas de vitamina D. Uso en la lactancia: No se sabe si Ácido Alendrónico/Colecalciferol se excreta en la leche materna humana. Sin embargo el Colecalciferol y algunos de sus metabolitos activos pasan a la leche materna. Alendronato/colecalciferol no se ha estudiado en madres lactantes y no debe administrarse a ellas.
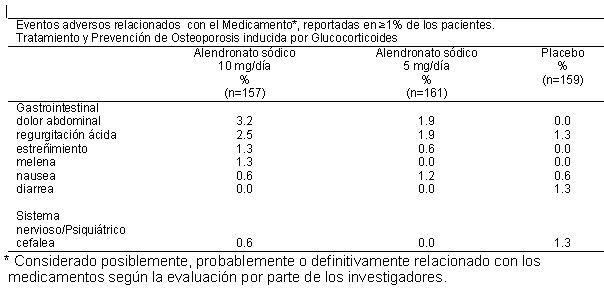
Reacciones secundarias y adversas: Reacciones adversas a medicamentos en ensayos clínicos. Debido a que los ensayos clínicos se llevan a cabo bajo condiciones muy específicas, las tasas de eventos adversos observados en los ensayos clínicos pueden no reflejar las tasas observadas en la práctica y no deben compararse con las tasas de los ensayos clínicos de otro medicamento. La información de los eventos adversos a los medicamentos obtenida de los ensayos clínicos es útil para identificar a aquellos que están relacionados con los medicamentos y estimar las tasas de frecuencia. Alendronato sódico: En los estudios clínicos, el alendronato sódico fue generalmente bien tolerado. En estudios con una duración de hasta cinco años, los eventos secundarios, que generalmente eran leves, por lo general no requirieron la interrupción del terapia. La seguridad del alendronato sódico se ha evaluado en estudios clínicos en aproximadamente 7,200 mujeres posmenopáusicas. Tratamiento de la Osteoporosis. Mujeres postmenopáusicas. En dos estudios multicéntricos, doble ciego, controlados con placebo con una duración de tres años (en Estados Unidos y Multinacionales) con un diseño prácticamente idéntico, en un total de 994 mujeres posmenopáusicas, los perfiles de seguridad general de alendronato sódico 10 mg/día y placebo fueron similares. La interrupción de la terapia debido a una experiencia clínica adversa se produjo en el 4.1% de 196 pacientes tratados con alendronato sódico 10 mg/día y 6.0% de 397 pacientes tratados con placebo. Los eventos adversos considerados por los investigadores como posiblemente, probablemente o definitivamente relacionadas en ≥ 1% de los pacientes tratados con alendronato sódico 10 mg/día o placebo se presentan en la siguiente tabla.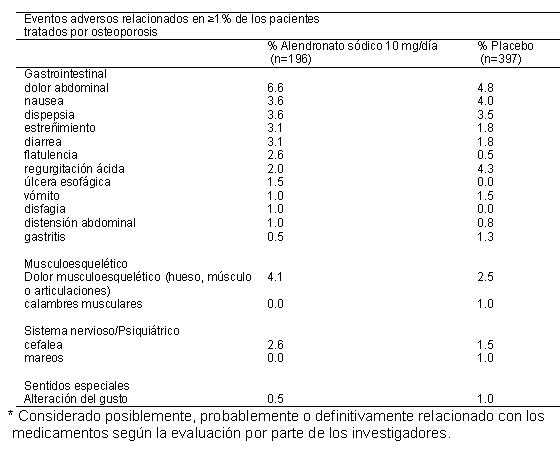
Un paciente tratado con alendronato sódico (10 mg/día) que tenía antecedentes de enfermedad ulcerosa péptica y gastrectomía y estaba tomando ácido acetilsalicílico concomitante (ASA), desarrolló una úlcera anastomótica con hemorragia leve, que se consideró relacionada con el medicamento. Tanto el ASA, como el alendronato sódico se suspendieron y el paciente se recuperó. En la extensión de dos años (años de tratamiento 4 y 5) de los estudios anteriores, el perfil de seguridad general de alendronato sódico 10 mg/día fue similar al que se observó durante el periodo controlado con placebo de tres años. Además, la proporción de pacientes que descontinuaron alendronato sódico 10 mg/día debido a una experiencia adversa clínica fue similar a aquella durante los primeros tres años del estudio. En el Ensayo sobre la Reducción de Fracturas (Fracture Intervention Trial), la interrupción del tratamiento debido a alguna experiencia adversa clínica se produjo en el 9.1% de 3,236 pacientes tratados con alendronato sódico 5 mg/día durante dos años y 10 mg/día por ya sea uno o dos años adicionales, y en 10.1% de 3,223 pacientes tratados con placebo. Las suspensiones del tratamiento debidas a eventos adversos gastrointestinales superiores fueron: alendronato sódico, 3.2%; placebo, 2.7%. El perfil general de eventos adversos fue similar al que se observó en otros estudios con alendronato sódico 5 o 10 mg/día. En un estudio multicéntrico doble ciego de un año, los perfiles de seguridad y tolerabilidad general de alendronato sódico 70 mg una vez por semana y alendronato sódico 10 mg una vez por día fueron similares. Los eventos adversos considerados por los investigadores como posiblemente, probablemente o definitivamente relacionadas con el fármaco en ≥ 1% de los pacientes en cualquiera de los grupos de tratamiento se presentan en la siguiente tabla: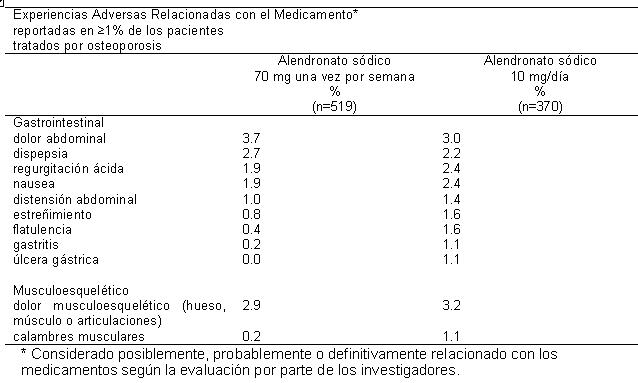
Hombres: En dos estudios doble ciego, multicéntricos, controlados contra placebo en hombres (estudio de dos años de duración con alendronato sódico 10 mg/día [n = 146] y un estudio de un año de alendronato sódico 70 mg una vez por semana [n = 109]), el perfil de seguridad de alendronato sódico fue generalmente similar al que se observó en mujeres posmenopáusicas. Las tasas de interrupción del tratamiento debido a alguna experiencia clínica adversa fueron de 2.7% para alendronato sódico 10 mg/día frente a 10.5% para placebo, y del 6.4% para alendronato sódico 70 mg una vez por semanal frente a 8.6% para placebo. Otros estudios realizados en hombres y mujeres: En un estudio de endoscopia de diez semanas de duración, realizado en hombres y mujeres (n = 277; edad media: 55), no se observaron diferencias en las lesiones del tracto gastrointestinal superior entre alendronato sódico 70 mg una vez por semana y placebo. En un estudio adicional de un año en hombres y mujeres (n = 335; edad media: 50), los perfiles generales de seguridad y tolerabilidad de alendronato sódico 70 mg una vez por semana fueron similares a los de placebo y no se observó ninguna diferencia entre hombres y mujeres. Otros estudios con alendronato sódico. Prevención de la osteoporosis en mujeres posmenopáusicas. La seguridad de alendronato sódico 5 mg/día en mujeres posmenopáusicas de 40-60 años de edad ha sido evaluada en tres estudios doble ciego, controlados contra placebo que incluyeron a más de 1,400 pacientes quienes fueron distribuidas e forma aleatoria para recibir alendronato sódico durante dos o tres años. En estos estudios, los perfiles generales de seguridad de alendronato sódico 5 mg/día y placebo fueron similares. La interrupción del tratamiento debido a cualquier experiencia clínica adversa se produjo en el 7.5% de 642 pacientes tratados con alendronato sódico 5 mg/día y el 57% de los 648 pacientes tratados con placebo. Los eventos adversos notificados por los investigadores como posible, probable o definitivamente relacionadas con el medicamento en ≥ 1% de los pacientes tratados con ya sea alendronato sódico 5 mg/día o placebo se presentan en la siguiente tabla: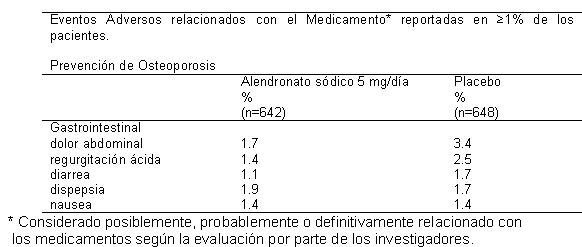
Uso concomitante con la terapia de reemplazo hormonal/estrógenos: En dos estudios (de uno y dos años de duración) en mujeres posmenopáusicas con osteoporosis (total: n = 853), los perfiles generales de seguridad y tolerabilidad del tratamiento combinado con alendronato sódico 10 mg una