
DAPOSAR
LIOMONT
Denominación genérica: Dapagliflozina
Forma farmacéutica y formulación: Tableta. Cada tableta contiene: Dapagliflozina 10 mg. Excipiente cbp 1 tableta.
Indicaciones terapéuticas: DAPOSAR® está indicado en monoterapia para mejorar el control glucémico en pacientes con diabetes mellitus tipo 2, como complemento a la dieta y el ejercicio, cuando no se considere adecuado el uso de metformina debido a intolerancia. DAPOSAR® está indicado como terapia complementaria para mejorar el control glucémico en pacientes con diabetes mellitus tipo 2, en combinación con metformina, una tiazolidinediona, una sulfonilurea (con o sin metformina), un inhibidor de DDP4 (con o sin metformina) o insulina (sola o en combinación con hasta dos antidiabéticos orales), cuando la terapia existente junto con la dieta y ejercicio no proporcionan un control glucémico adecuado. DAPOSAR® está indicado como terapia de combinación inicial con metformina, para mejorar el control glucémico en pacientes con diabetes mellitus tipo 2 cuando es apropiada la terapia dual de dapagliflozina y metformina, como complemento a la dieta y el ejercicio.
Farmacocinética y farmacodinamia: Farmacocinética: La exposición a dapagliflozina se incrementó de forma proporcional al aumento de la dosis de dapagliflozina en el intervalo de 0.1 a 500 mg y su farmacocinética no cambió con el tiempo con la administración diaria repetida durante hasta 24 semanas. Absorción: La dapagliflozina se absorbe bien y con rapidez después de la administración oral. Las concentraciones plasmáticas máximas (Cmáx) de dapagliflozina, se alcanzaron normalmente en las 2 horas siguientes a la administración en ayunas. Los valores de Cmáx y exposición sistémica a dapagliflozina (ABC) se incrementaron en forma proporcional al incremento en la dosis de dapagliflozina. La biodisponibilidad oral absoluta de dapagliflozina, después de la administración de una dosis de 10 mg, es de 78%. Los alimentos tuvieron un efecto relativamente modesto en la farmacocinética de dapagliflozina en sujetos sanos. La administración con una comida rica en grasas disminuyó la Cmáx de dapagliflozina en hasta 50% y prolongó el Tmáx en aproximadamente 1 hora, pero no alteró el ABC en comparación con la administración en ayunas. Se consideró que estos cambios no fueron clínicamente significativos. Por lo tanto, la dapagliflozina puede administrarse con o sin alimentos. Distribución: La dapagliflozina se une a las proteínas en un 91% aproximadamente. La unión a las proteínas no se modificó en presencia de diversas enfermedades (por ejemplo, insuficiencia renal o hepática). El volumen de distribución en estado estacionario de dapagliflozina fue de 118 L. Metabolismo: La dapagliflozina se metaboliza en forma extensa y produce principalmente dapagliflozina 3-O-glucurónido, un metabolito inactivo. Ni dapagliflozina 3-O-glucurónido ni otros metabolitos contribuyen a los efectos hipoglucemiantes. La formación de dapagliflozina 3-O-glucurónido está medida por la UGT1A9, una enzima presente en el hígado y el riñón, y el metabolismo mediado por CYP es una vía de eliminación secundaria en humanos. La dapagliflozina consta de un enlace glucosídico, lo que significa que el componente aglicona está unido a la glucosa mediante un enlace carbono-carbono, lo que le confiere estabilidad contra enzimas glucosidasas. La vida media terminal plasmática (t½) para dapagliflozina es de 12.9 horas después de una dosis oral única de 10 mg en sujetos sanos. La dapagliflozina 3-O-glucurónido representó 61% de una dosis de 50 mg de (14C)-dapagliflozina, siendo el componente predominante relacionado con el fármaco en el plasma humano que representa 42% (basado en el ABC [0-12 h]) de la radiactividad plasmática total, similar a la contribución de 39% del fármaco original. Basado en el ABC, ningún otro metabolito representó > 5% de la radiactividad plasmática total en cualquier tiempo medido. Eliminación: La dapagliflozina y los metabolitos relacionados se eliminan fundamentalmente mediante excreción urinaria, de la cual menos del 2% es dapagliflozina intacta. Después de la administración de una dosis de 50 mg de (14C)-dapagliflozina, se recuperó el 96%, el 75% en la orina y el 21% en las heces. En heces, aproximadamente 15% de la dosis se excretó como fármaco original. Poblaciones especiales: No se recomiendan ajustes de dosis con base en análisis farmacocinéticos para insuficiencia renal leve, insuficiencia hepática leve, moderada y severa, edad, género, raza y peso corporal. Insuficiencia renal: La dapagliflozina no debe ser usado en pacientes con insuficiencia renal moderada o severa (tasa de filtración glomerular estimada [TFGe] persistente < 45 mL/min/1.73 m2 o depuración de creatinina [DepCr] persistente < 60 mL/min). En estado estacionario (dapagliflozina 20 mg una vez al día durante 7 días), los pacientes con diabetes mellitus tipo 2 e insuficiencia renal leve, moderada y severa (determinado por depuración de iohexol), tuvieron exposiciones sistémicas promedio de dapagliflozina que fueron un 32%, 60% y 87% superiores, respectivamente, a las observadas en pacientes con diabetes mellitus tipo 2 y función renal normal. Con 20 mg de dapagliflozina una vez al día, la exposición sistémica más alta a dapagliflozina en pacientes con diabetes mellitus tipo 2 e insuficiencia renal, no resultó en una depuración renal más elevada de glucosa o excreción de glucosa en 24 horas. La depuración renal de glucosa y la excreción de glucosa de 24 horas fue inferior en pacientes con insuficiencia renal moderada o severa en comparación con pacientes con insuficiencia renal normal y leve. La excreción urinaria de glucosa en 24 horas fue altamente dependiente de la función renal, 85, 52, 18 y 11 g de glucosa/día se excretaron en pacientes con diabetes mellitus tipo 2 y función renal normal o insuficiencia renal leve, moderada o severa, respectivamente. No hubo diferencias en la unión a proteínas de dapagliflozina entre los grupos de insuficiencia renal o en comparación con sujetos sanos. No se conoce el efecto de la hemodiálisis sobre la exposición a dapagliflozina. Insuficiencia hepática: En los pacientes con insuficiencia hepática leve o moderada, la Cmáx y el ABC promedio de dapagliflozina fue hasta de 12 y 36% superiores, respectivamente, en comparación con los sujetos control sanos. Estas diferencias no se consideraron clínicamente importantes y no se propone ajuste de dosis a partir de la dosis usual propuesta de 10 mg una vez al día de dapagliflozina para estas poblaciones. En los pacientes con insuficiencia hepática severa (clase Child-Pugh C), la Cmáx y el ABC promedio de dapagliflozina fueron de hasta 40% y 67% superiores, que en los sujetos control sanos, respectivamente. No se requiere ajuste de dosis en pacientes con insuficiencia hepática severa. Sin embargo, el riesgo-beneficio del uso de dapagliflozina en pacientes con insuficiencia hepática severa, se debería valorar en forma individual puesto que la seguridad y eficacia de dapagliflozina no se ha estudiado específicamente en esta población. Edad avanzada: No hay un aumento clínicamente significativo en la exposición en función de la edad en sujetos de hasta 70 años de edad. Sin embargo, cabe esperar un aumento en la exposición debido a la disminución en la función renal relacionada con la edad. No hay suficientes datos sobre la exposición en pacientes mayores de 70 años de edad. Pacientes pediátricos y adolescentes: No se ha estudiado la farmacocinética en la población pediátrica y adolescente. Sexo: No se recomienda ajuste de dosis para dapagliflozina a partir de la dosis de 10 mg una vez al día con base en el género. El ABC promedio de dapagliflozina en mujeres (n = 619) se estima 22% más elevada que en hombres (n = 634) (IC 90%: 117, 124). Raza: No se recomienda ajuste de dosis para dapagliflozina a partir de la dosis de 10 mg una vez al día con base en la raza. No hay diferencias clínicamente significativas en las exposiciones sistémicas entre las razas blanca, negra o asiática. Peso corporal: No se recomienda ajuste de dosis para dapagliflozina a partir de la dosis propuesta de 10 mg una vez al día con base en el peso. Se ha observado que la exposición a dapagliflozina disminuye con el aumento de peso. En consecuencia, los pacientes con bajo peso corporal pueden presentar una exposición ligeramente aumentada y pacientes con pesos elevados una exposición ligeramente disminuida. Sin embargo, las diferencias en la exposición no se consideraron clínicamente significativas. Farmacodinamia: La Dapagliflozina es un potente y altamente selectivo inhibidor oral del cotransportador de sodio-glucosa tipo 2 (SGLT2) en humanos, el principal transportador responsable de la reabsorción de glucosa renal. La dapagliflozina es un inhibidor selectivo altamente potente y reversible del cotransportador de sodio-glucosa tipo 2 (SGLT2) que mejora el control glucémico en pacientes con diabetes mellitus tipo 2 mediante la reducción de la reabsorción de glucosa del filtrado glomerular en el túbulo proximal renal con una reducción concomitante de la reabsorción de sodio que conduce a la excreción urinaria de glucosa (glucosuria) y a la diuresis osmótica. La dapagliflozina está disponible en forma oral y requiere de la administración una vez al día. El SGLT2 se expresa en forma selectiva en el riñón sin expresión detectada en más de otros 70 tejidos incluyendo hígado, músculo esquelético, tejido adiposo, mama, vejiga y cerebro. El SGLT2 es el transportador predominante responsable de la reabsorción de la glucosa proveniente del filtrado glomerular que regresa a la circulación. A pesar de la presencia de hiperglucemia en la diabetes mellitus tipo 2, la reabsorción de glucosa filtrada continúa. La dapagliflozina reduce el transporte máximo de glucosa tubular en 55% y reduce la reabsorción renal de glucosa, de tal manera que la glucosa aparece en la orina a niveles normales de glucosa plasmática. Por lo tanto, la dapagliflozina mejora los niveles plasmáticos de glucosa en ayuno y posprandial reduciendo la reabsorción renal de glucosa, lo que conduce a la excreción urinaria del exceso de glucosa. Esta excreción de glucosa (efecto glucosúrico) se observa después de la primera dosis, es continua durante el intervalo de administración de 24 horas y se mantiene durante el tratamiento. La cantidad de glucosa eliminada por el riñón mediante este mecanismo depende de la concentración de glucosa en la sangre y de la tasa de filtración glomerular (GFR). Por tanto, en sujetos sanos con glucosa sanguínea normal, la dapagliflozina tiene una baja propensión a causar hipoglucemia. La dapagliflozina no altera la producción normal endógena de glucosa en respuesta a la hipoglucemia. La dapagliflozina actúa en forma independiente de la secreción de insulina y de la acción de la insulina. Con el tiempo, se ha observado mejoría en la función de las células beta (HOMA-2) en estudios clínicos con dapagliflozina. La excreción urinaria de glucosa (glucosuria), inducida por dapagliflozina, está asociada con una pérdida calórica y reducción en el peso. La mayoría de la reducción en el peso fue pérdida de grasa corporal, incluyendo la grasa visceral, más que masa magra o pérdida de volumen, como se demuestra por absorciometría de rayos X de energía dual (DXA) e imágenes de resonancia magnética. La inhibición del cotransporte de glucosa y sodio por parte de dapagliflozina también se asocia con diuresis leve y natriuresis transitoria. La dapagliflozina no inhibe otros transportadores de glucosa importantes para el transporte de glucosa hacia tejidos periféricos y es 1,400 veces más selectiva para SGLT2 frente a SGLT1, el principal transportador en el intestino responsable de la absorción de glucosa. Efectos farmacodinámicos: Se observaron aumentos de la cantidad de glucosa excretada en la orina en sujetos sanos y en sujetos con diabetes mellitus tipo 2 después de la administración de dapagliflozina. Se excretaron aproximadamente 70 g de glucosa en la orina por día (equivalente a 280 kcal/día), con una dosis de dapagliflozina de 10 mg/día en pacientes con diabetes mellitus tipo 2 durante 12 semanas. Se observó evidencia de excreción sostenida de glucosa en pacientes con diabetes mellitus tipo 2 que recibieron dapagliflozina 10 mg/día hasta por 2 años. Esta excreción urinaria de glucosa con dapagliflozina también provoca diuresis osmótica y aumenta el volumen urinario en los sujetos con diabetes mellitus tipo 2. El incremento en el volumen urinario en pacientes con diabetes mellitus tipo 2 tratados con dapagliflozina de 10 mg se mantuvieron a las 12 semanas y representaron aproximadamente 375 mL/día. El aumento del volumen urinario se asoció con un aumento pequeño y transitorio en la excreción urinaria de sodio que no se asoció con cambios en las concentraciones séricas de sodio. La excreción urinaria de ácido úrico también aumentó en forma transitoria (durante 3-7 días) y estuvo acompañada por una reducción en la concentración sérica de ácido úrico. A las 24 semanas, las reducciones en las concentraciones séricas de ácido úrico fueron de -0.33 mg/dL a -0.87 mg/dL. Electrofisiología cardiaca: La dapagliflozina no se asoció con la prolongación clínicamente significativa del intervalo QTc en dosis diarias de hasta 150 mg (15 veces la dosis recomendada) en un estudio de sujetos sanos. Además, no se observó ningún efecto clínicamente importante en el intervalo QTc después de dosis únicas de hasta 500 mg (50 veces la dosis recomendada) de dapagliflozina en sujetos sanos.
Contraindicaciones: DAPOSAR® está contraindicado en pacientes con antecedentes de cualquier reacción de hipersensibilidad a la dapagliflozina o a cualquiera de los excipientes de este medicamento. DAPOSAR® está contraindicado en diabetes mellitus tipo 1, cetoacidosis diabética, insuficiencia renal crónica, pacientes menores de 18 años, embarazo y lactancia.
Precauciones generales: Uso en pacientes con insuficiencia renal. DAPOSAR® no se deberá usar en pacientes con insuficiencia renal moderada a severa (TFGe persistente < 45 mL/min/1.73 m2 o DepCr persistente < 60 mL/min). Por lo tanto, al igual que en todos los pacientes con diabetes, se deberá evaluar la función renal antes de iniciar el tratamiento con DAPOSAR®, así como periódicamente después del inicio. La eficacia hipoglucemiante de DAPOSAR® depende de la función renal. DAPOSAR® no se ha estudiado en pacientes con insuficiencia renal severa (TFGe < 30 mL/min/1.73 m2 o DepCr ≤30 mL/ min) o enfermedad renal terminal (ERT) y, por lo tanto, no se debe usar en esta población. Uso en pacientes con riesgo de depleción de volumen o hipotensión El efecto diurético de DAPOSAR® disminuye el volumen intravascular que a veces puede manifestarse como hipotensión sintomática o cambios transitorios agudos en la creatinina. Por lo tanto, se debe tener precaución en pacientes para los que una caída de la presión arterial inducida por dapagliflozina pudiera suponer un riesgo, tales como pacientes con tratamiento antihipertensivo, con antecedentes de hipotensión o insuficiencia renal y pacientes de edad avanzada. En caso de enfermedades intercurrentes que puedan conducir a una depleción del volumen (por ejemplo, enfermedades gastrointestinales), se recomienda un estrecho monitoreo del estado del volumen (por ejemplo, exploración física, medición de la presión arterial, estudios de laboratorio). Se recomienda la interrupción temporal del tratamiento con DAPOSAR® en pacientes que desarrollen depleción del volumen hasta que este problema se corrija. Antes de iniciar DAPOSAR® en estos pacientes, se debe evaluar el estado del volumen y la función renal. Cetoacidosis: Se han notificado casos raros de cetoacidosis diabética durante la poscomercialización, incluyendo cetoacidosis diabética en pacientes con diabetes mellitus tipo 1 y 2 que están tomando dapagliflozina y otros inhibidores de SGLT2, aunque no se ha establecido la relación causal. DAPOSAR® no está indicado para el tratamiento de pacientes con diabetes mellitus tipo 1. No se recomienda reiniciar el tratamiento con el inhibidor del SGLT2 en pacientes que experimenten cetoacidosis diabética durante el tratamiento con un inhibidor del SGLT2, a menos que se identifique otro factor bien definido que lo desencadenara y se haya resuelto. El riesgo de cetoacidosis diabética aún si los niveles de glucosa en sangre están por debajo de 14 mmol/L (250 mg/dL), se debe considerar en el caso de síntomas inespecíficos tales como náuseas, vómitos, anorexia, dolor abdominal, malestar general, sed excesiva, dificultad respiratoria, confusión, fatiga o somnolenica inusuales. Si estos síntomas aparecen, se debe evaluar de forma inmediata a los pacientes para valorar si se trata de una cetoacidosis, independientemente de los niveles de glucosa en sangre. Si se sospecha de cetoacidosis, debe considerarse la suspensión o interrupción temporal de DAPOSAR®. Se debe interrumpir el tratamiento en pacientes que están hospitalizados por un procedimiento quirúrgico mayor o enfermedades agudas graves. Se recomienda controlar las cetonas en estos pacientes. Se prefiere la determinación de los niveles de cuerpos cetónicos en sangre a la determinación en orina. El tratamiento con dapagliflozina se puede reanudar cuando los valores de cuerpos cetónicos sean normales y el estado del paciente se haya estabilizado. Antes de iniciar DAPOSAR® se deben considerar los factores en la historia clínica del paciente que predispongan a la cetoacidosis. Los factores que predisponen a la cetoacidosis incluyen un bajo funcionamiento de las células beta resultante de desórdenes pancreáticos (por ejemplo, diabetes tipo 1, historia de pancreatitis o cirugía pancreática), reducción de la dosis de insulina, ingesta calórica reducida o el incremento de requerimientos de insulina debido a infecciones, enfermedad o cirugía y abuso de alcohol. DAPOSAR® debe ser usado con precaución en estos pacientes. Uso en pacientes en estado de hiperosmolaridad: DAPOSAR® no debe ser usado en pacientes en estado hiperosmolar. Fascitis necrosante del perineo (gangrena de Fournier): Se han notificado casos poscomercialización de fascitis necrosante del perineo (también conocida como gangrena de Fournier) en pacientes de ambos sexos tratados con inhibidores del SGLT2. Se trata de un acontecimiento raro pero grave y potencialmente mortal que requiere intervención quirúrgica urgente y tratamiento antibiótico. Se indicará a los pacientes que acudan al médico si presentan una combinación de síntomas como dolor, dolor a la palpación, eritema o inflamación en la región genital o perineal, con fiebre o malestar general. Tenga en cuenta que la infección urogenital o el absceso perineal pueden preceder a la fascitis necrosante. Si se sospecha gangrena de Fournier, se debe interrumpir DAPOSAR® e instaurar un tratamiento inmediato (incluidos antibióticos y desbridamiento quirúrgico). Uso con medicamentos que causan hipoglucemia: La insulina y los secretagogos de la insulina, como sulfonilureas causan hipoglucemia. Por lo tanto, se podría requerir de una dosis menor de insulina o del secretagogo de la insulina para reducir el riesgo de hipoglucemia cuando se usa en combinación con DAPOSAR®. Infecciones del tracto urinario: Se han presentado reportes poscomercialización de infecciones serias en vías urinarias, incluyendo urosepsis y pielonefritis, que requieren hospitalización en pacientes que reciben dapagliflozina y otros inhibidores SGLT2. El tratamiento con inhibidores SGLT2 incrementa el riesgo de infecciones en vías urinarias. Si se indica DAPOSAR®, los pacientes deben ser evaluados por signos y síntomas de infecciones en el tracto urinario y tratados de inmediato. Infecciones micóticas genitales: DAPOSAR® aumenta el riesgo de infecciones micóticas genitales. Los pacientes con antecedentes de infecciones micóticas genitales tienen más probabilidades de desarrollar infecciones micóticas geniales, por lo tanto, se recomienda vigilar a estos pacientes y tratar adecuadamente. Uso en población geriátrica: No se recomiendan cambios de dosis de DAPOSAR® basados en la edad. Los pacientes de edad avanzada pueden presentar mayor riesgo de depleción del volumen y es más probable que sean tratados con diuréticos. Es más probable que los pacientes de edad avanzada presenten una función renal alterada, y/o estén en tratamiento con medicamentos antihipertensivos que puedan provocar cambios en la función renal tales como inhibidores de la enzima conversora de la angiotensina (IECA) y antagonistas del receptor tipo 1 de la angiotensina II (ARA). Se aplican las mismas recomendaciones para la función renal en pacientes de edad avanzada, que para los demás pacientes. Resultados macrovasculares: No se han reportado estudios clínicos en los que se establezca evidencia concluyente de la reducción del riesgo macrovascular con dapagliflozina o con algún otro antidiabético. En un metaanálisis de 21 estudios clínicos, el uso de dapagliflozina no se asoció con un incremento en el riesgo de eventos adversos cardiovasculares. Uso en pacientes con diabetes y enfermedad cardiovascular: El tratamiento con dapagliflozina de 10 mg con terapia de adición a tratamientos antidiabéticos preexistentes por más de 24 semanas proporcionó mejorías significativas de HbA1c y beneficio clínico comparado con placebo en esta población. También se observaron reducciones significativas en el peso corporal total y en la presión arterial sistólica en posición sentada. Estos beneficios se extendieron hasta la semana 104 de tratamiento. Análisis de orina: Debido a su mecanismo de acción, los pacientes que estén tomando DAPOSAR®, presentarán resultados positivos para la glucosa en orina. Efectos en la capacidad para conducir u operar maquinaria: No se han realizado estudios sobre los efectos en la capacidad para conducir u operar maquinaria.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: DAPOSAR® no se debe usar en el segundo y tercer trimestre del embarazo, el cual corresponde al periodo de la maduración renal humana. Los estudios realizados en ratas han mostrado toxicidad para el riñón en desarrollo, incremento en la incidencia o severidad de dilataciones renales pélvicas y tubulares en la progenie en el periodo de tiempo correspondiente al segundo y tercer trimestre del embarazo humano. Debido a que no hay estudios adecuados y bien controlados de dapagliflozina en mujeres embarazadas, cuando se detecta un embarazo, debe suspenderse el tratamiento con DAPOSAR®. Lactancia: DAPOSAR® no debe usarse durante la lactancia. Los datos farmacodinámicos/toxicológicos disponibles en animales muestran excreción de dapagliflozina o metabolitos en la leche, así como efectos farmacológicos en las crías lactantes. Se desconoce si dapagliflozina o su metabolito se excretan en la leche humana.
Reacciones secundarias y adversas: Diabetes mellitus tipo 2: En los estudios clínicos en diabetes tipo 2, más de 15,000 pacientes han sido tratados con dapagliflozina. La evaluación primaria de seguridad y tolerabilidad se llevó a cabo en un análisis conjunto pre-especificado de 13 estudios a corto plazo (hasta 24 semanas) controlados con placebo con 2,360 sujetos tratados con dapagliflozina 10 mg y 2,295 tratados con placebo. En un estudio de resultados cardiovasculares de dapagliflozina en diabetes mellitus tipo 2 (estudio DECLARE) 8,574 pacientes recibieron dapagliflozina 10 mg y 8,569 recibieron placebo durante una mediana de tiempo de exposición de 48 meses. En total, hubo 30,623 pacientes-año de exposición a dapagliflozina. Las reacciones adversas notificadas con más frecuencia a través de los estudios clínicos fueron infecciones genitales. Insuficiencia cardiaca: En el estudio de resultados cardiovasculares de dapagliflozina en pacientes con insuficiencia cardiaca con fracción de eyección reducida (estudio DAPA-HF), se trato a 2,368 pacientes con dapagliflozina 10 mg y 2,368 con placebo durante una mediana de tiempo de exposición de 18 meses. La población de pacientes incluía pacientes con diabetes mellitus tipo 2 y sin diabetes, y pacientes con TFGe ≥30 mL/min/1.73 m2. En el estudio de resultados cardiovasculares de dapagliflozina en pacientes con insuficiencia cardiaca con fracción de eyección del ventrículo izquierdo > 40% (DELIVER), se trato a 3,126 pacientes con dapagliflozina 10 mg y 3,127 pacientes con placebo durante una medida de tiempo de exposición de 27 meses. La población de pacientes incluía pacientes con diabetes mellitus tipo 2 y sin diabetes, y pacientes con TFGe ≥25 mL/min/1.73 m2. El perfil de seguridad general de dapagliflozina en los pacientes con insuficiencia cardiaca fue consistente con el perfil de seguridad conocido de dapagliflozina. Enfermedad renal crónica: En el estudio de resultados renales de dapagliflozina en pacientes con enfermedad renal crónica (DAPA-CKD), se trato a 2,149 pacientes con placebo durante una mediana de tiempo de 27 meses. La población de pacientes incluía pacientes con diabetes mellitus tipo 2 y sin diabetes, con TFGe ≥ 25 a ≤75 mL/min/1.73 m2, y albuminuria (cociente de albúmina/creatinina en orina [CACo] ≥200 y ≤5, 000 mg/g) continuaba si la TFGe disminuía a niveles por debajo de 25 mL/min/1.73 m2. El perfil de seguridad general de dapagliflozina en pacientes con enfermedad renal crónica fue consistente con el perfil de seguridad conocido de dapagliflozina. Resumen tabulado de reacciones adversas: Las reacciones adversas han sido ordenadas según frecuencias utilizando la siguiente clasificación: Muy frecuentes ( > 1/10); frecuentes ( > 1/100 a < 1/10); poco frecuentes ( > 1/1.000 a < 1/100); raras ( > 1/10.000 a < 1/1.000); muy raras ( < 1/10.000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles. Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada intervalo de frecuencia.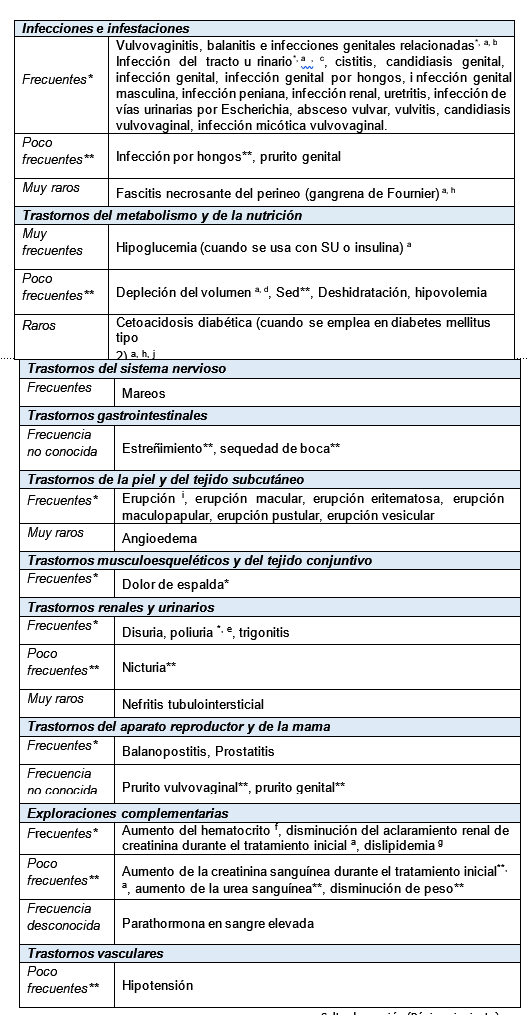
a Ver información adicional a continuación en la subsección correspondiente. b Vulvovaginitis, balanitis e infecciones genitales relacionadas incluyen, por ejemplo, los términos preferentes predefinidos: Infección micótica vulvovaginal, infección vaginal, balanitis, infección fúngica genital, candidiasis vulvovaginal, vulvovaginitis, balanitis por Candida, candidiasis genital, infección genital, infección genital masculina, infección del pene, vulvitis, vaginitis bacteriana y absceso vulvar. c Infección del tracto urinario incluye los siguientes términos preferentes, listados en orden de frecuencia notificada: Infección del tracto urinario, cistitis, infección del tracto urinario por Escherichia, infección del tracto geniturinario, pielonefritis, trigonitis, uretritis, infección renal y prostatitis. d La depleción del volumen incluye, por ejemplo, los términos preferentes predefinidos: Deshidratación, hipovolemia, hipotensión. e La poliuria incluye los siguientes términos preferentes: Polaquiuria, poliuria, aumento de la diuresis. f La variación media del hematocrito respecto del valor inicial fue del 2,30% con dapagliflozina 10 mg frente al -0,33% con placebo. Los valores de hematocrito > 55% fueron notificados en el 1,3% de los sujetos tratados con dapagliflozina 10 mg frente al 0,4% de los sujetos con placebo. g El porcentaje medio de cambio desde el valor inicial para dapagliflozina 10 mg frente a placebo, respectivamente, fue de: Colesterol total 2,5% frente a -0,0%; colesterol HDL 6,0% frente a 2,7%; colesterol LDL 2,9% frente a -1,0%; triglicéridos -2,7% frente a -0,7%. h Ver sección precauciones generales. i La reacción adversa fue identificada durante la vigilancia poscomercialización. Erupción incluye los siguientes términos preferentes, listados en orden de frecuencia en los estudios clínicos: Erupción, erupción generalizada, erupción prurítica, erupción macular, erupción maculo-papular, erupción pustular, erupción vesicular y erupción eritematosa. En estudios clínicos controlados con activo o con placebo (dapagliflozina, N=5936, Control total, N=3403), la frecuencia de la erupción fue similar en dapagliflozina (1,4%) y en el control total (1,4%), respectivamente. j Notificada en el estudio de resultados cardiovasculares en pacientes con diabetes tipo 2 (DECLARE). La frecuencia se basa en el promedio anual. * Notificadas en ≥ 2% de los sujetos y ≥ 1% más y al menos 3 sujetos más tratados con dapagliflozina 10 mg en comparación con placebo. **Notificadas por el investigador como posiblemente relacionadas, probablemente relacionadas o relacionadas con el tratamiento del ensayo y notificadas en ≥ 0,2% de los sujetos y un ≥ 0,1% más veces y en al menos 3 sujetos más tratados con dapagliflozina 10 mg en comparación con placebo. Descripción de reacciones adversas seleccionadas: Vulvovaginitis, balanitis e infecciones genitales relacionadas: En el conjunto de los 13 estudios de seguridad, se notificó vulvovaginitis, balanitis e infecciones genitales relacionadas en el 5.5% y 0.6% de los sujetos que recibieron dapagliflozina 10 mg y placebo, respectivamente. La mayoría de las infecciones fueron de leves a moderadas y los sujetos respondieron a un ciclo inicial de tratamiento convencional y rara vez ocasionaron la interrupción del tratamiento con dapagliflozina. Estas infecciones fueron más frecuentes en mujeres (8.4% y 1.2% para dapagliflozina y placebo, respectivamente) y los sujetos con antecedentes presentaban mayor probabilidad de infección recurrente. En el estudio DECLARE, el número de pacientes con acontecimientos adversos graves de infecciones genitales fue bajo y equilibrado: 2 pacientes en cada uno de los grupos de dapagliflozina y placebo. En el estudio DAPA-HF, ningún paciente informó de acontecimientos adversos graves por infecciones genitales en el grupo de dapagliflozina y uno en el grupo de placebo. Hubo 7 (0.3%) pacientes con acontecimientos adversos que dieron lugar a interrupciones por infecciones genitales en el grupo de dapagliflozina y ninguno en el grupo de placebo. En el estudio DELIVER, 1 paciente ( < 0.1%) en cada grupo de tratamiento notificó un acontecimiento adverso grave de infecciones genitales. Hubo 3 pacientes (0.1%) con acontecimientos adversos que dieron lugar a interrupciones por infecciones genitales en el grupo de dapagliflozina y ninguno en el grupo de placebo. En el estudio DAPA-CKD, hubo 3 (0.1%) pacientes con acontecimientos adversos graves por infecciones genitales en el grupo de dapagliflozina y ninguno en el grupo de placebo. Hubo 3 (0.1%) pacientes con acontecimientos adversos que dieron lugar a la interrupción debido a infecciones genitales en el grupo de dapagliflozina y ninguno en el grupo de placebo. Los acontecimientos adversos graves de infecciones genitales o acontecimientos adversos que dieron lugar a la interrupción debido a infecciones genitales no se notificaron para ningún paciente sin diabetes. Fascitis necrosante del perineo (gangrena de Fournier): Se han notificado casos poscomercialización de gangrena de Fournier en pacientes tratados con inhibidores SGLT2, incluyendo dapagliflozina. En el estudio DECLARE con 17,160 pacientes con diabetes mellitus tipo 2 y una mediana de tiempo de exposición de 48 meses, se reportaron un total de 6 casos de gangrena de Fournier, uno en el grupo tratado con dapagliflozina y 5 en el grupo con placebo. Hipoglucemia: La frecuencia de hipoglucemia dependió del tipo de tratamiento de base utilizado en los estudios clínicos en la diabetes mellitus. Para estudios de dapagliflozina en monoterapia, de adición a metformina o de adición a sitagliptina (con o sin metformina), la frecuencia de episodios menores de hipoglucemia fue similar ( < 5%) entre los grupos de tratamiento, incluido el grupo placebo hasta las 102 semanas de tratamiento. En todos los estudios, los acontecimientos mayores de hipoglucemia fueron poco frecuentes y comparables entre los grupos tratados con dapagliflozina o placebo. Los estudios con tratamientos de adición a sulfonilurea y de adición a insulina presentaron mayor incidencia de hipoglucemia. En un estudio de adición a glimepirida, en las semanas 24 y 48, se notificaron episodios menores de hipoglucemia más frecuentemente en el grupo tratado con dapagliflozina 10 mg más glimepirida (6.0% y 7.9%, respectivamente) que en el grupo de placebo más glimepirida (2.1% y 2.1%, respectivamente). En un estudio de adición a insulina se notificaron episodios de hipoglucemia grave en el 0.5% y 1.0% de los sujetos tratados con dapagliflozina 10 mg más insulina en las semanas 24 y 104, respectivamente, y en el 0.5% de los sujetos de grupos tratados con placebo más insulina en las semanas 24 y 104. En las semanas 24 y 104, se notificaron episodios de hipoglucemia leve, respectivamente, en el 40.3% y 53.1% de los sujetos que recibieron dapagliflozina 10 mg más insulina y en el 34.0% y 41,6% de los sujetos que recibieron placebo más insulina. En un estudio de adición a metformina y una sulfonilurea, de hasta 24 semanas, no se notificaron episodios de hipoglucemia grave. Se notificaron episodios menores de hipoglucemia en el 12.8% de los sujetos que recibieron dapagliflozina 10 mg más metformina y una sulfonilurea y en el 3.7% de los que recibieron placebo más metformina y una sulfonilurea. En el estudio DECLARE, no se observó un aumento del riesgo de hipoglucemia grave con la terapia de dapagliflozina en comparación con placebo. Se notificaron acontecimientos graves de hipoglucemia en 58 (0.7%) pacientes tratados con dapagliflozina y en 83 (1.0%) pacientes tratados con placebo. En el estudio DAPA-HF se notificaron acontecimientos graves de hipoglucemia en 4 (0.2%) pacientes de ambos grupos de tratamiento, dapagliflozina y placebo; y se observaron sólo en pacientes con diabetes mellitus tipo 2. En el estudio DAPA-CKD, los acontecimientos graves de hipoglucemia fueron notificados en 14 (0.7%) pacientes en el grupo de dapagliflozina y 28 (1.3%) pacientes en el grupo de placebo. En el estudio DELIVER, se notificaron acontecimientos graves de hipoglucemia en 6 (0.2%) pacientes en el grupo de dapagliflozina y 7 (0.2%) en el grupo de placebo. Los acontecimientos graves de hipoglucemia solo se observaron en pacientes con diabetes mellitus tipo 2. Depleción del volumen: En el conjunto de los 13 estudios de seguridad, se notificaron reacciones indicativas de depleción del volumen (incluyendo notificaciones de deshidratación, hipovolemia o hipotensión) en el 1.1% y 0.7%, de los sujetos tratados con dapagliflozina 10 mg, y placebo, respectivamente; las reacciones graves se dieron en < 0.2% de los sujetos, repartidos de forma equilibrada entre dapagliflozina 10 mg y placebo. En el estudio DECLARE, el número de pacientes con acontecimientos indicativos de depleción del volumen fue equilibrado entre los grupos de tratamiento: 213 (2,5%) y 207 (2,4%) en los grupos de dapagliflozina y placebo, respectivamente. Se notificaron acontecimientos adversos graves en 81 (0.9%) y 70 (0.8%) en los grupos de dapagliflozina y placebo, respectivamente. Los acontecimientos fueron generalmente equilibrados entre los grupos de tratamiento a través de los subgrupos de edad, uso de diurético, presión arterial e inhibidores de la enzima convertidora de angiotensina (IECA)/antagonistas de los receptores tipo 1 de angiotensina II (ARAII). En pacientes con valores iniciales de TFGe < 60 ml/min/1,73 m2, hubo 19 casos de reacciones adversas graves compatibles con depleción del volumen en el grupo de dapagliflozina y 13 casos en el grupo placebo. En el estudio DAPA-HF, el número de pacientes con acontecimientos sugestivos de una depleción del volumen fue de 170 (7.2%) en el grupo de dapagliflozina y de 153 (6.5%) en el grupo placebo. Hubo menos pacientes con acontecimientos graves de síntomas sugestivos de una depleción del volumen en el grupo de dapagliflozina (23 [1.0%]) en comparación con el grupo placebo (38 [1.6%]). Los resultados fueron similares independientemente de la presencia basal de diabetes y de la TFGe inicial. En el estudio DAPA-CKD, el número de pacientes con acontecimientos sugestivos de depleción de volumen fue de 120 (5.6%) en el grupo de dapagliflozina y 84 (3.9%) en el grupo placebo. Hubo 16 (0.7%) pacientes con acontecimientos graves de síntomas sugestivos de depleción de volumen en el grupo de dapagliflozina y 15 (0.7%) pacientes en el grupo de placebo. En el estudio DELIVER, el número de pacientes con acontecimientos graves de síntomas sugestivos una depleción de volumen fue de 35 (1.1%) en el grupo de dapagliflozina y 31 (1.0%) en el grupo de placebo. Cetoacidosis diabética (CAD) en diabetes mellitus tipo 2: En el estudio DECLARE, con una mediana de tiempo de exposición de 48 meses, los acontecimientos de CAD se notificaron en 27 pacientes en el grupo de dapagliflozina 10 mg y 12 pacientes en el grupo placebo. Los acontecimientos ocurrieron distribuidos de forma uniforme durante el periodo del estudio. De los 27 pacientes con acontecimientos de CAD en el grupo de dapagliflozina, 22 tomaban insulina como tratamiento concomitante al mismo tiempo que el acontecimiento. Los factores desencadenantes de CAD fueron los esperados en una población de diabetes mellitus tipo 2. En el estudio DAPA-HF, se notificaron acontecimientos de CAD en 3 pacientes con diabetes mellitus tipo 2 en el grupo de dapagliflozina y ninguno en el grupo placebo. En el estudio DELIVER, se notificaron acontecimientos de CAD en 2 pacientes con diabetes mellitus tipo 2 en el grupo de dapagliflozina y ninguno en el grupo de placebo. En el estudio DAPA-CKD, no se notificaron acontecimientos de CAD en ningún paciente en el grupo de dapagliflozina y en 2 pacientes con diabetes mellitus tipo 2 en el grupo de placebo. Infecciones del tracto urinario: En el conjunto de los 13 estudios de seguridad, las infecciones del tracto urinario se notificaron más frecuentemente con dapagliflozina 10 mg en comparación con placebo (4,7% frente al 3,5%, respectivamente; ver sección precauciones generales). La mayoría de las infecciones fueron de leve a moderadas, y los sujetos respondieron a un ciclo inicial de tratamiento convencional y rara vez ocasionaron la interrupción del tratamiento con dapagliflozina. Estas infecciones fueron más frecuentes en mujeres y los sujetos con antecedentes presentaban mayor probabilidad de infección recurrente. En el estudio DECLARE, los acontecimientos graves de infecciones del tracto urinario se notificaron de forma menos frecuente para dapagliflozina 10 mg en comparación con placebo, 79 (0.9%) acontecimientos frente a 109 (1.3%) acontecimientos, respectivamente. En el estudio DAPA-HF, el número de pacientes con acontecimientos adversos graves de infecciones del tracto urinario fue de 14 (0.6%) en el grupo de dapagliflozina y 17 (0.7%) en el grupo placebo. Hubo 5 (0.2%) pacientes con acontecimientos adversos que dieron lugar a interrupciones por infecciones del tracto urinario en cada uno de los grupos, dapagliflozina y placebo. En el estudio DELIVER el número de pacientes con eventos adversos graves de infecciones del tracto urinario fue 41 (1.3%) en el grupo dapagliflozina 37 (1.2%) y en el grupo placebo hubo 13 (0.4%) pacientes con eventos adversos que llevaron a discontinuaciones debido a infecciones del tracto urinario en el grupo de dapagliflozina y 9 (0.3%) en el grupo placebo. En el estudio DAPA-CKD, el número de pacientes con acontecimientos adversos graves de infecciones del tracto urinario fue de 29 (1.3%) en el grupo de da