DARITEX-A
FRESENIUS KABI
Denominación genérica: Irinotecan.
Forma farmacéutica y formulación: Solución. El frasco ámpula contiene: Clorhidrato de irinotecan trihidratado 40 mg y 100 mg. Vehículo cbp 2 ml y 5 ml.
Indicaciones terapéuticas: Antineoplásico para el tratamiento de cáncer colorrectal metastásico, gástrico, ovario, pulmón en células pequeñas y no pequeñas, cervical.
Farmacocinética y farmacodinamia: Farmacocinética: Después de la infusión intravenosa en humanos, las concentraciones en plasma del Irinotecan declinan en una manera multiexponencial, con una vida media de eliminación terminal de aproximadamente 6 horas. La vida media de eliminación terminal del metabolito activo SN-38 es de aproximadamente 10 horas. Las vidas medias de las formas de lactona activa del irinotecan y del SN-38 son similares a las de los compuestos primarios, las formas de lactona y de hidroxiácido se encuentran en equilibrio. Sobre el intervalo de dosificación de 50 a 350 mg/m², el área bajo la curva (ABC) de irinotecan se incrementa linealmente con la dosis; el ABC del SN-38 se incrementa menos que proporcionalmente con la dosis. Las concentraciones máximas del metabolito activo SN-38 son generalmente observadas después de una hora posterior al final de una infusión de 90 minutos de irinotecan. El irinotecan muestra una unión moderada a las proteínas del plasma (30-68%); mientras que el SN-38 se une altamente a las proteínas plasmáticas humanas (aproximadamente 95%). La proteína plasmática a la cual se unen principalmente el irinotecan y el SN-38 es la albúmina.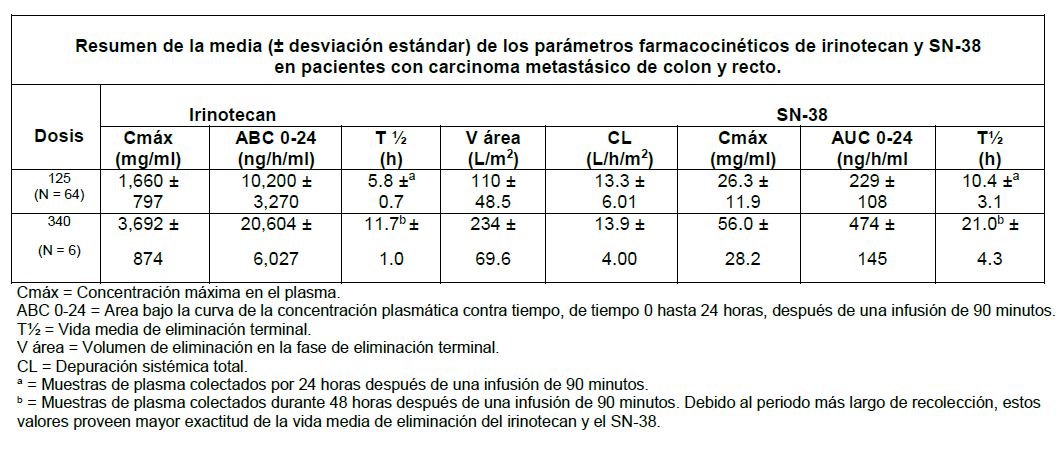
Metabolismo y excreción: La conversión metabólica del irinotecan al metabolito activo SN-38 está mediada por enzimas carboxilesterasas y ocurre primariamente en el hígado. El SN-38 subsecuentemente sufre una conjugación para formar un metabolito glucurónido. El SN-38 glucurónido tiene 1/50 a 1/100 de actividad del SN-38 en ensayos de citotoxicidad utilizando líneas celulares para estudios in vitro. La disposición del irinotecan no ha sido completamente elucidada en humanos. La excreción urinaria del irinotecan es del 11% al 20%; SN-38, < 1%; y SN-38 glucurónido, 3%. La excreción acumulativa biliar y urinaria de irinotecan y sus metabolitos sobre un periodo de 48 horas después de la administración de irinotecan en dos pacientes, se encuentra en el intervalo de aproximadamente el 25% (100 mg/m2) a 50% (300 mg/m2). Farmacocinéticas en subpoblaciones: Geriátricas: La vida media terminal del irinotecan fue de 6.0 horas en pacientes de 65 o más años y de 5.5 horas en pacientes más jóvenes de 65 años. La ABC O-24 para SN-38 en pacientes que tenían al menos 65 años de edad fue 11% más alta que en pacientes menores de 65 años. No se recomiendan cambios en la dosis y administración para pacientes geriátricos. Pediátricos: Las farmacocinéticas de irinotecan no han sido estudiadas en poblaciones pediátricas. Género: Las farmacocinéticas del irinotecan no parecen estar influenciadas por el género. Raza: La influencia de la raza en las farmacocinéticas del irinotecan no ha sido evaluada. Insuficiencia hepática: La influencia de la insuficiencia hepática en las características farmacocinéticas del irinotecan y sus metabolitos no ha sido formalmente estudiada. En pacientes con tumor hepático conocido (una mayoría de los pacientes), los valores del ABC del irinotecan y el SN-38 fueron algo más altos que los valores para pacientes sin metástasis en el hígado. Para pacientes que tienen metástasis en el hígado sin disminución en la función hepática, no se recomiendan cambios en la dosis o la administración. Insuficiencia renal: La influencia de la insuficiencia renal en las farmacocinéticas del irinotecan no han sido evaluadas. Farmacodinamia: DARITEX-A es un agente antineoplásico de la clase de los inhibidores de la topoisomerasa I, clínicamente investigado como CPT-11. El irinotecan es un derivado semisintético de la campotecina, un alcaloide extraído de plantas como Camptotheca acuminata. Mecanismo de acción: Las camptotecinas interactúan específicamente con la enzima topoisomerasa I, la cual está implicada en la síntesis del ADN. El irinotecan y su metabolito activo SN-38 se unen al complejo de la topoisomerasa I-ADN y previenen la re-unión de la banda simple. Las investigaciones actuales sugieren que la citotoxicidad del irinotecan es debida al daño que produce en la doble cadena durante la síntesis del ADN, cuando las enzimas de replicación interactúan con el complejo ternario formado por la topoisomerasa I, ADN y también irinotecan o SN-38. Las células mamíferas no pueden reparar eficientemente estos rompimientos en la doble hélice. El irinotecan sirve como un precursor soluble en agua del metabolito lipofílico SN-38. El SN-38 es formado a partir del irinotecan por la carboxiesterasa, mediante la separación de la unión entre la porción de camptotecina y la cadena lateral de fipiperidino. El SN-38 es aproximadamente 1,000 veces más potente que el irinotecan como un inhibidor de la topoisomerasa I purificado de líneas celulares de tumores de humanos y roedores. Los ensayos de citotoxicidad in vitro muestran que la potencia del SN-38 relacionada con el irinotecan varía de 2 a 2,000 veces; el SN-38 se une aproximadamente en un 95% a las proteínas del plasma, mientras que el irinotecan sólo se une en un 50%. La contribución precisa del SN-38 al irinotecan es todavía desconocida. Ambos existen en forma de lactona activa y de un anión hidroxiácido. Un equilibrio dependiente de pH existe entre las dos formas, de manera que el pH ácido promueve la formación de lactona, mientras que un pH más básico favorece la formación de un anión hidroxiácido.
Contraindicaciones: Pacientes con hipersensibilidad conocida a la sustancia activa o a sus aditivos. Así mismo, no deberá utilizarse durante el embarazo y la lactancia.
Precauciones generales: Administración: El irinotecan solamente se debe administrar bajo la supervisión de un médico con experiencia en el uso de agentes quimioterapéuticos para el tratamiento del cáncer. Solamente es posible el manejo adecuado de las complicaciones cuando se dispone fácilmente del diagnóstico adecuado y de las instalaciones para el tratamiento. Síntomas colinérgicos: Los pacientes pueden tener síntomas colinérgicos de rinitis, salivación aumentada, miosis, lagrimeo, diaforesis, enrojecimiento (vasodilatación), bradicardia e hiperperistaltismo intestinal que puede causar calambres abdominales y diarrea temprana (es decir, la diarrea que generalmente ocurre durante o dentro de 8 horas de la administración del irinotecan). Se cree que estos síntomas, los cuales se pueden observar durante o poco después de la infusión del irinotecan, están relacionados con la actividad anticolinérgica del irinotecan inalterado, y se espera que ocurran más frecuentemente con dosis mayores de irinotecan. En los pacientes con síntomas colinérgicos se debe considerar la administración terapéutica o profiláctica de 0.25 a 1 mg de atropina intravenosa o subcutánea (a menos que esté contraindicada). Extravasación: Se debe tener cuidado de evitar la extravasación, y se debe vigilar el sitio de la infusión para detectar signos de inflamación. Si ocurre extravasación, se recomienda lavar el sitio y aplicar hielo. Hepáticas: En los estudios clínicos se han observado anormalidades de las enzimas hepáticas grado 3 ó 4 del National Cancer Institute (NCI) en menos del 10% de los pacientes. Típicamente estos eventos ocurren en los pacientes con metástasis hepáticas conocidas, y no están claramente relacionadas con el irinotecan. Hematológicas: Comúnmente el irinotecan causa neutropenia, leucopenia y anemia, los cuales pueden ser severos. La trombocitopenia severa es poco común. En los estudios clínicos, la frecuencia de neutropenia grado 3 y 4 del NCI ha sido significativamente mayor en los pacientes que recibieron irradiación pélvica/abdominal previa que en los pacientes que no habían sido expuestos a esta irradiación. Los pacientes con niveles basales de bilirrubina sérica total de 1.0 mg/dl o más, también tuvieron una probabilidad significativamente mayor de neutropenia grado 3 ó 4 en el primer ciclo de la terapia que los pacientes con niveles de bilirrubina por debajo de 1.0 mg/dl. No hubo diferencias significativas entre la edad y el sexo en la frecuencia de neutropenia grado 3 ó 4. En los estudios clínicos, la fiebre neutropénica (neutropenia recurrente grado 4 del NCI y fiebre grado 2) ocurrió en menos del 10% de los pacientes; sin embargo, en los pacientes tratados con irinotecan se han reportado muertes por sepsis después de mielosupresión severa. Si ocurre fiebre neutropénica, o si la cuenta absoluta de neutrófilos cae por debajo de 1,000/mm3, suspender la terapia con irinotecan temporalmente. La dosis de irinotecan se debe disminuir si ocurre mielosupresión clínicamente significativa (ver Dosis y vía de administración). Reacciones de hipersensibilidad: Se han reportado reacciones de hipersensibilidad, incluyendo reacciones anafilácticas/anafilactoides severas. Diarrea tardía: La diarrea tardía (que generalmente ocurre después de más de ocho horas de la administración del irinotecan) puede ser prolongada, causar deshidratación y desequilibrio electrolítico, y poner en peligro la vida. En los estudios clínicos que utilizaron el régimen de dosificación cada tres semanas, el tiempo mediano para el inicio de la diarrea tardía fue de 5 días después de la infusión del irinotecan. En los estudios clínicos para evaluar el esquema de dosificación semanal, el tiempo mediano para el inicio de la diarrea tardía fue de 11 días después de la administración del irinotecan. Para los pacientes que empezaron el tratamiento con una dosis de 125 mg/m2 semanalmente, y que experimentaron diarrea tardía grado 3 ó 4, la duración mediana de todo el episodio de diarrea fue de 7 días. La frecuencia por edad de la diarrea tardía grado 3 y 4 fue significativamente mayor en los pacientes de 65 años de edad o mayores que en los pacientes menores de 65 años de edad. En asociación con la diarrea inducida por el irinotecan se ha observado ulceración del colon, algunas veces con sangrado. La diarrea tardía se debe tratar inmediatamente con loperamida al primer episodio de heces poco formadas o sueltas, o al primer inicio de movimientos intestinales más frecuentes que los normalmente esperados para el paciente. El régimen de dosificación recomendado para loperamida es de 4 mg en el primer inicio de la diarrea tardía y posteriormente 2 mg cada 2 horas hasta que el paciente esté libre de diarrea por lo menos 12 horas. Durante la noche, el paciente puede tomar 4 mg de loperamida cada 4 horas. No se recomienda la premedicación con loperamida. Los pacientes con diarrea severa deben ser vigilados cuidadosamente, y se les debe administrar líquidos y reemplazo de electrólitos, si se deshidratan. Si ocurre diarrea grado 2, 3 ó 4 del NCI, se debe suspender la administración del irinotecan, o se debe disminuir la dosis (ver Dosis y vía de administración, Recomendaciones para la modificación de la dosis). Náuseas y vómito: El irinotecan es emetogénico. La náusea y el vómito pueden ser severos, y usualmente ocurren durante o poco después de la infusión del irinotecan. Se recomienda que los pacientes reciban premedicación con agentes antieméticos. Los agentes antieméticos se administrarán en el día del tratamiento, empezando por lo menos 30 minutos antes de la administración del irinotecan. Además, los médicos deben considerar prescribir a sus pacientes un régimen antiemético para uso subsiguiente según se requiera. Neurológicas: Se ha observado mareo, y algunas veces puede representar evidencia sintomática de hipotensión ortostática en los pacientes con deshidratación. Renales: Se han observado aumentos en los niveles de creatinina en suero o de urea en sangre. Generalmente estos eventos se han atribuido a la deshidratación relacionada con náusea, vómito o diarrea. También se han reportado raros casos de disfunción renal debida al síndrome de lisis tumoral. Respiratorias: Se ha observado disnea grado 3 ó 4 del NCI. No se sabe en qué magnitud el compromiso pulmonar u otra enfermedad preexistente puede contribuir a la disnea. En los primeros estudios realizados en Japón, en un pequeño porcentaje de pacientes se observó un síndrome pulmonar que potencialmente pone en peligro la vida, consistente en disnea, fiebre y un patrón reticulonodular en la radiografía de tórax. Fue difícil evaluar la contribución del irinotecan en estos eventos preliminares porque los pacientes tenían además tumores pulmonares y alguna enfermedad pulmonar no maligna preexistente. Poblaciones especiales: Niños: No se ha establecido la seguridad y efectividad del irinotecan en los pacientes pediátricos. Ancianos: Los médicos deben tener precaución al tratar a pacientes de 65 a 70 años de edad o mayores, debido a un mayor riesgo de diarrea en esta población. Se pueden aplicar las recomendaciones de dosificación específicas para esta población, dependiendo del régimen usado (ver Dosis y vía de administración). Terapia de irradiación: Los pacientes que previamente han recibido irradiación pélvica/abdominal tienen un mayor riesgo de mielosupresión después de la administración del irinotecan. Los médicos deben tener precaución al tratar a pacientes con irradiación extensa previa. Se pueden aplicar las recomendaciones de dosificación específicas para esta población, dependiendo del régimen usado (ver Dosis y vía de administración). Condición física de ejecución (actividades-ECOG): Los pacientes con una condición física pobre tienen un mayor riesgo de eventos adversos relacionados con el irinotecan. Se pueden aplicar las recomendaciones de dosificación específicas para los pacientes con un estado de condición física de 2 del Eastern Cooperative Oncology Group (ECOG), dependiendo del régimen usado (ver Dosis y vía de administración). Los pacientes con un estado de condición física de 3 ó 4 no deben ser tratados con irinotecan. Cáncer gástrico: Los pacientes con cáncer gástrico parecen experimentar mayor mielosupresión y otras toxicidades cuando son tratados con irinotecan. En estos pacientes se debe considerar una dosis inicial menor (ver Dosis y vía de administración).
Restricciones de uso durante el embarazo y la lactancia: El irinotecan es teratogénico en ratas y conejos. El irinotecan puede causar daño fetal cuando se administra a una mujer embarazada. No se han realizado estudios adecuados y bien controlados con irinotecan en mujeres embarazadas. Si el fármaco se usa durante el embarazo, o si la paciente se embaraza mientras está recibiendo este fármaco, debe ser advertida del peligro potencial para el feto. Las mujeres con probabilidades de embarazo deben ser advertidas de evitar embarazarse mientras reciben tratamiento con irinotecan. En ratas se detectó radiactividad en la leche dentro de 5 minutos de la administración intravenosa de irinotecan marcado radiactivamente y se concentró hasta 65 veces a las 4 horas después de la administración, en comparación con las concentraciones plasmáticas. Como muchos fármacos se excretan en la leche humana y dado el potencial de reacciones adversas severas en los lactantes, se recomienda interrumpir la lactancia cuando se recibe terapia con irinotecan.
Reacciones secundarias y adversas: Estudios clínicos: Se han recolectado y analizado exhaustivamente los datos de los eventos adversos, obtenidos en el programa de estudios clínicos de cáncer colorrectal metastásico recurrente o progresivo después de terapia con 5-FU (segunda línea), y se presentan a continuación (la población de los pacientes se describe más adelante). Se espera que los eventos adversos para otras indicaciones sean similares a los eventos adversos de la terapia de segunda línea para el cáncer colorrectal. Estudios clínicos de dosis únicas de 100 a 125 mg/m2 del fármaco sólo administrado semanalmente: El esquema de dosificación semanal de irinotecan se evaluó en tres estudios clínicos en 304 pacientes con carcinoma metastásico del colon o recto que había recurrido o progresado después de la terapia con 5-FU. Cinco muertes (1.6%) fueron consideradas potencialmente relacionadas con el fármaco. Estos cinco pacientes experimentaron una variedad de eventos médicos (mielosupresión, sepsis neutropénica sin fiebre, obstrucción del intestino delgado, acumulación de líquido, estomatitis, náusea, vómito, diarrea y deshidratación), los cuales son efectos conocidos del irinotecan. La fiebre neutropénica, definida como neutropenia grado 4 del NCI y fiebre grado 2 o mayor, ocurrió en otros 9 pacientes; éstos se recuperaron con el cuidado de soporte. Ochenta y un pacientes (26.6%) fueron hospitalizados debido a eventos considerados como relacionados con la administración de irinotecan. Las razones principales para la hospitalización relacionada con el fármaco fueron diarrea, con o sin náuseas y/o vómito; neutropenia/leucopenia, con o sin diarrea y/o fiebre; y náuseas y/o vómito. Durante el ciclo de tratamiento y en los ciclos subsiguientes se realizaron ajustes en las dosis de irinotecan, con base en la tolerancia individual del paciente. Las razones más comunes para la disminución de la dosis fueron la diarrea tardía, neutropenia y leucopenia. Trece pacientes (4.3%) suspendieron el tratamiento con irinotecan debido a eventos adversos. Estudio clínico del esquema de dosificación con 300 a 350 mg/m2 una vez cada tres semanas, usado como fármaco solo: Un total de 316 pacientes con cáncer colorrectal metastásico cuya enfermedad había progresado después de terapia previa con 5-FU recibieron irinotecan en dos estudios que comprendieron la administración del fármaco una vez cada tres semanas. Tres muertes (1%) fueron consideradas potencialmente relacionadas con el irinotecan, y se atribuyeron a infección neutropénica, diarrea grado 4, y astenia, respectivamente. Las hospitalizaciones debidas a eventos adversos severos, relacionados o no con la administración de irinotecan, ocurrieron en por lo menos 60% de los pacientes que recibieron irinotecan, y 8% de los pacientes tratados con irinotecan interrumpieron el tratamiento debido a eventos adversos. Listas de los eventos adversos: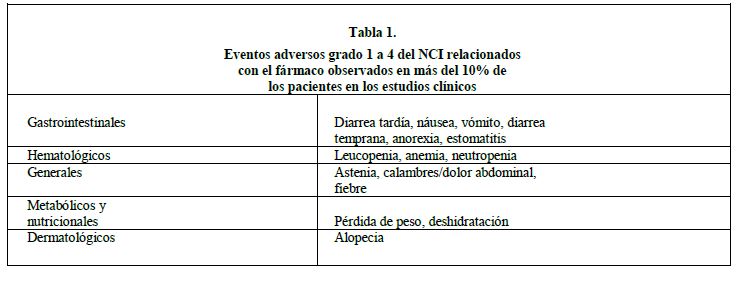
En las tablas 2 a 4 se enlistan los eventos adversos severos grados 3 ó 4 del NCI reportados en los estudios clínicos de los esquemas de dosificación semanalmente y una vez cada 3 semanas (N = 620).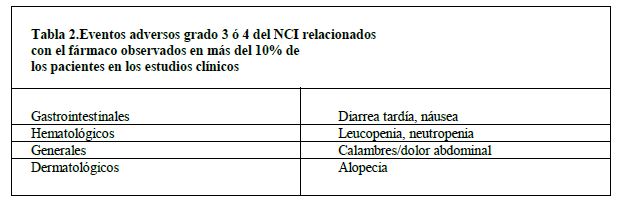
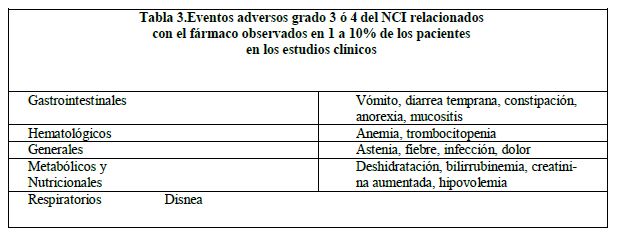
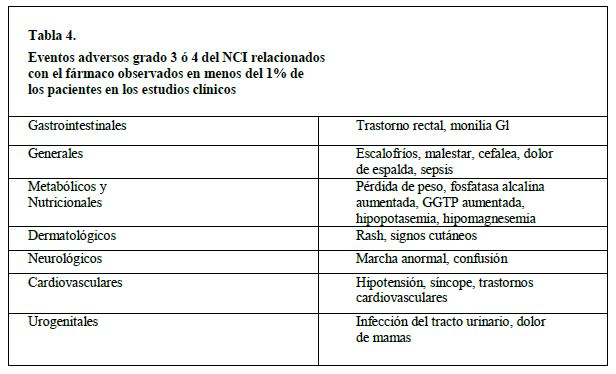
En los estudios clínicos del irinotecan se han reportado los siguientes eventos adversos relacionados con fármaco, pero no cumplen con los criterios definidos anteriormente, ya sea de evento adverso grado 1-4 del NCI relacionado con el fármaco en más del 10% de los pacientes, o como un evento adverso grado 3 ó 4 relacionado con el fármaco: rinitis, salivación aumentada, miosis, lagrimeo, diaforesis, enrojecimiento, bradicardia, mareo, extravasación, síndrome de lisis tumoral y ulceración del colon. Vigilancia post-comercialización: Se han reportado casos de colitis. En algunos casos, la colitis estuvo complicada por cólico o megacolon tóxico. También se han reportado casos de cólico sin colitis precedente. Se han reportado reacciones de hipersensibilidad, incluyendo reacciones anafilácticas o anafilactoides severas.
Interacciones medicamentosas y de otro género: Agentes antineoplásicos: Se espera que los efectos adversos del irinotecan, como mielosupresión y diarrea, sean exacerbados por otros agentes antineoplásicos con un perfil similar de efectos adversos. Dexametasona: Se ha reportado linfocitopenia en los pacientes tratados con irinotecan, y es posible que la administración de dexametasona como profilaxis antiemética pueda tener una mayor probabilidad de linfocitopenia. Sin embargo, no se han observado infecciones oportunistas, y no se ha atribuido ninguna complicación específicamente a la linfocitopenia. Se ha observado hiperglucemia en los pacientes con historia de diabetes mellitus o evidencia de intolerancia a la glucosa antes de la administración del irinotecan. Es posible que en algunos pacientes la dexametasona, administrada como profilaxis antiemética, contribuya a la hiperglucemia. Laxantes: Se espera que el uso de laxantes durante la terapia empeore la incidencia o severidad de la diarrea. Diuréticos: El irinotecan puede inducir deshidratación secundaria a vómito y/o diarrea. El médico puede suspender los diuréticos durante el tratamiento con irinotecan y durante los periodos de vómito y diarrea activos.
Alteraciones en los resultados de pruebas de laboratorio: No existen interacciones conocidas entre las pruebas de laboratorio y el irinotecan; sin embargo, se recomienda la monitorización cuidadosa de la cuenta de leucocitos, hemoglobina y cuenta plaquetaria antes de cada dosis de DARITEX-A.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: No se han llevado a cabo estudios a largo plazo con irinotecan. Sin embargo, se han administrado dosis intravenosas a las ratas de 2 mg/kg o 25 mg/kg de irinotecan una vez por semana por 13 semanas (en estudios separados, la dosis de 25 mg/kg produjo una Cmáx de irinotecan y una ABC 7 veces y 1.3 veces los valores respectivos en pacientes a los que se les administró 125 mg/m²) y se recuperaron después de 91 semanas. Bajo estas condiciones una tendencia lineal significativa correlacionada con la dosis para la incidencia de pólipos endometriales o uterino y sarcomas estromales endometriales. Ni el irinotecan ni el SN-38 fueron mutagénicos en los ensayos in vitro. Sin embargo, en estudios in vitro en células de hámster chinos, el irinotecan produjo un incremento significativo en la incidencia de aberraciones cromosomales dependiente de la concentración. Adicionalmente, en un ensayo in vivo con ratones, una sola dosis intraperitoneal de irinotecan sobre el intervalo de la dosis de 2.5 a 200 mg/kg causó un incremento significativo dependiente de la dosis en la formación de eritrocitos policromáticos micronucleados, y una disminución en la proporción de reticulocitos/eritrocitos en las células de la médula ósea. Reproducción: No se han observado efectos adversos significativos en la fertilidad y la reproductividad general después de la administración intravenosa de irinotecan en dosis de hasta 6.0 mg/kg/día en ratas. Sin embargo, la atrofia de los órganos reproductores masculinos se ha observado después de la administración de dosis múltiples diarias de irinotecan, tanto en roedores como en perros. La radiactividad relacionada con el irinotecan marcado con C14 cruza la placenta de las ratas después de la administración intravenosa de 10 mg/kg, que corresponden a valores de 125 mg/m2 administrados en pacientes, y en conejos a 6 mg/kg/día (aproximadamente la mitad de la dosis humana recomendada semanalmente). Los efectos teratogénicos incluyen una variedad de efectos externos, viscerales y esqueletales. El irinotecan administrado en ratas madres para el periodo seguido a la organogénesis a través de la lactancia a dosis de 6 mg/kg/día causó disminución en la capacidad de aprendizaje y disminuyó los pesos corporales de las hembras en el nacimiento.
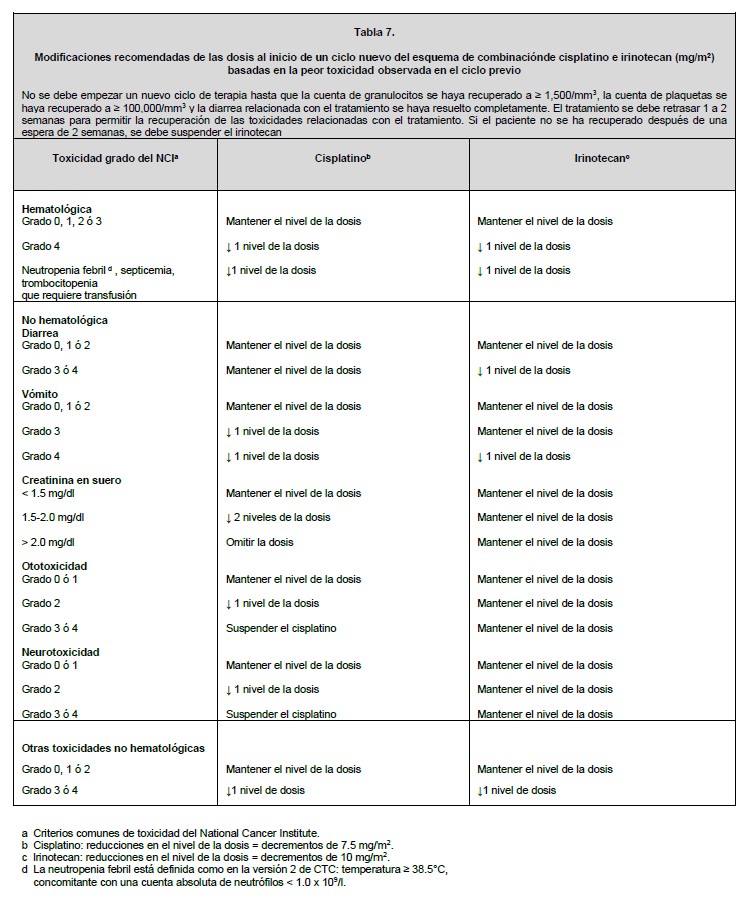
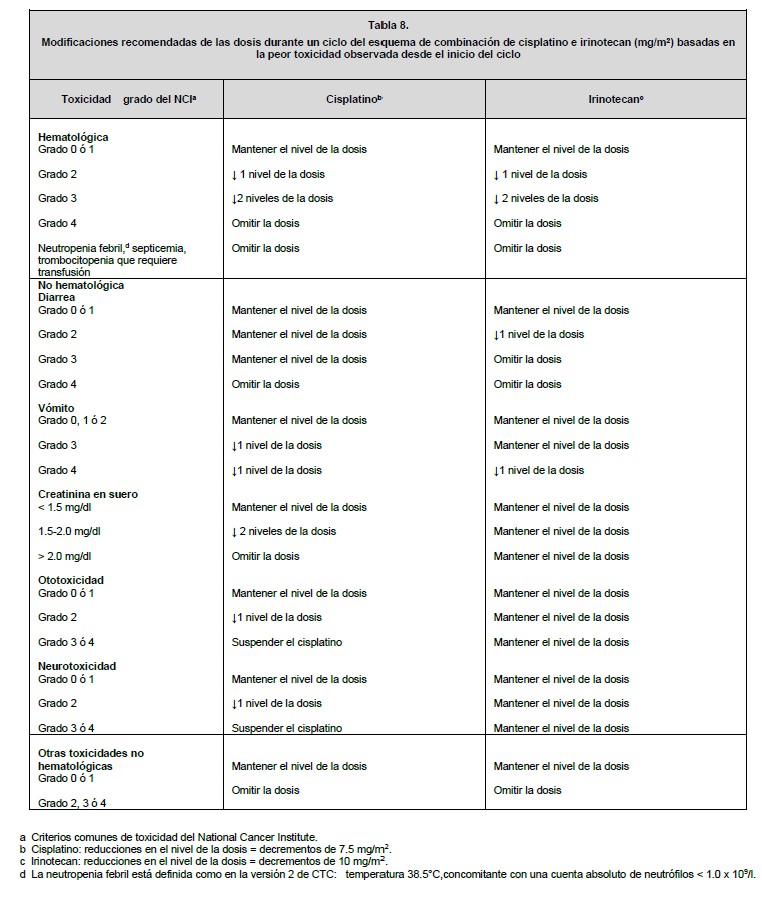
Dosis y vía de administración: La vía de administración es parenteral exclusivamente infusión intravenosa. Todas las dosis de irinotecan se deben administrar como una infusión intravenosa durante 30 a 90 minutos. Preparación: Antes de la infusión, el irinotecan se debe diluir en dextrosa al 5% para inyección, o en cloruro de sodio al 0.9% para inyección, hasta una concentración final en el intervalo de 0.12 a 2.8 mg/ml. Manejo: Al igual que con otros agentes anticancerígenos potencialmente tóxicos, se debe tener cuidado en el manejo y preparación de las soluciones de irinotecan preparadas para infusión. Se recomienda usar guantes. Si el irinotecan entra en contacto con la piel, lavar inmediata y abundantemente con jabón y agua. Si el irinotecan entra en contacto con las mucosas, lavar abundantemente con agua. Los productos farmacéuticos parenterales se deben inspeccionar visualmente para detectar materia particulada y decoloración antes de su administración, siempre y cuando la solución y el contenedor lo permitan. Inspeccionar el contenido del vial para detectar materia particulada y repetir la inspección al tomar el producto del vial con la jeringa. Mezclas diluidas: La solución es física y químicamente estable hasta 24 horas a temperatura ambiente y luz fluorescente ambiental. Las soluciones diluidas en dextrosa al 5% para inyección, almacenadas en refrigeración y protegidas de la luz, son física y químicamente estables durante 48 horas. No se recomienda la refrigeración de las mezclas preparadas con cloruro de sodio al 0.9% para inyección, debido a una baja y esporádica incidencia de partículas visibles. Debido a la posible contaminación durante la dilución, es aconsejable usar la mezcla dentro de 24 horas en refrigeración, o dentro de 6 horas si se mantiene a temperatura ambiente. Se debe evitar la congelación de los viales de irinotecan o de las mezclas de irinotecan, en vista de que puede causar la precipitación del activo. Incompatibilidades: No se deben agregar otros fármacos en la solución para infusión. Esquemas de dosificación del irinotecan solo: Se han estudiado ampliamente los regímenes de dosificación de irinotecan solo para el cáncer colorrectal metastásico. Estos regímenes se pueden usar en el tratamiento de pacientes con otros cánceres indicados (ver Indicaciones terapéuticas). Dosis inicial: Esquema de dosificación semanal: La dosis inicial recomendada de irinotecan solo es de 125 mg/m2. Se puede considerar una dosis menor (por ejemplo, 100 mg/m2 para los pacientes con cualquiera de las siguientes condiciones: edad de 65 años o más, radioterapia extensa previa, estado de condición física de 2, niveles aumentados de bilirrubina, o cáncer gástrico. El tratamiento se debe administrar en ciclos repetidos de 6 semanas, que comprenden el tratamiento semanal durante 4 semanas, seguido por un descanso de 2 semanas. Esquema de dosificación una vez cada 2 semanas: La dosis inicial usual recomendada de irinotecan es de 250 mg/m2 cada 2 semanas mediante infusión intravenosa. Se puede considerar una dosis inicial menor (por ejemplo, 200 mg/m2) para los pacientes con cualquiera de las siguientes condiciones: 65 años de edad o más, radioterapia extensa previa, estado de condición física de 2, niveles aumentados de bilirrubina, o cáncer gástrico. Esquema de dosificación una vez cada 3 semanas: La dosis inicial usual recomendada de irinotecan para el esquema de dosificación una vez cada 3 semanas, es de 350 mg/m2. Se puede considerar una dosis inicial menor (por ejemplo, 300 mg/m2) para los pacientes con cualquiera de las siguientes condiciones: 65 años de edad o más, radioterapia extensa previa, estado de condición física de 2, niveles aumentados de bilirrubina, o cáncer gástrico. Esquema de dosificación de irinotecan en combinación: Dosis inicial: Irinotecan en combinación con 5-fluorouracilo (5-FU) y leucovorina: Se recomienda el uso de irinotecan en combinación con 5-FU y leucovorina en los pacientes con cáncer colorrectal metastásico. La dosis inicial recomendada es de 125 mg/m2 de irinotecan, 500 mg/m2 de 5-FU y 20 mg/m2 de leucovorina. Se pueden considerar dosis iniciales menores de irinotecan (por ejemplo, 100 mg/m2) y 5-FU (por ejemplo, 400 mg/m2) para los pacientes con cualquiera de las siguientes condiciones: 65 años de edad y mayores, radioterapia extensa previa, estado de condición física de 2, niveles aumentados de bilirrubina, o cáncer gástrico. El tratamiento se debe administrar en ciclos repetidos de 6 semanas, que comprenden el tratamiento semanal durante 4 semanas, seguido por un descanso de 2 semanas. Irinotecan en combinación con cisplatino: El irinotecan se ha estudiado en combinación con cisplatino para el cáncer pulmonar de células no pequeñas y de células pequeñas, cáncer cervical, cáncer gástrico y cáncer esofágico. Este régimen se puede usar en el tratamiento de otros cánceres indicados, excepto para el cáncer colorrectal (ver Indicaciones terapéuticas). La dosis inicial recomendada es de 65 mg/m2 de irinotecan y 30 mg/m2 de cisplatino. Se puede considerar una dosis inicial menor de irinotecan (por ejemplo, 50 mg/m2) para los pacientes con cualquiera de las siguientes condiciones: 65 años de edad o más, radioterapia extensa previa, estado de condición física de 2, niveles aumentados de bilirrubina, o cáncer gástrico. El tratamiento se debe administrar en ciclos repetidos de 6 semanas, comprendiendo el tratamiento semanal durante 4 semanas, seguido por un descanso de 2 semanas. Duración del tratamiento: Para los regímenes de irinotecan solo y en combinación, se puede continuar indefinidamente el tratamiento con ciclos adicionales de irinotecan en los pacientes que logran una respuesta tumoral, o en los pacientes cuyo cáncer permanece estable. Los pacientes deben ser vigilados cuidadosamente para detectar toxicidad, y se debe suspender la terapia si ocurre toxicidad inaceptable que no responda a la modificación de la dosis y al cuidado rutinario de soporte. Recomendaciones para la modificación de la dosis: En la tabla 5 se describen las modificaciones recomendadas de las dosis durante un ciclo de terapia y al inicio de cada ciclo subsiguiente de terapia con irinotecan solo. Estas recomendaciones se basan en las toxicidades comúnmente observadas con la administración de irinotecan. Para las modificaciones al inicio de un ciclo subsiguiente de terapia, la dosis de irinotecan se debe disminuir con relación a la dosis inicial del ciclo previo. En la tabla 6 se describen las modificaciones recomendadas de las dosis de irinotecan y 5-FU durante un ciclo de terapia y al inicio de cada ciclo subsiguiente de terapia con irinotecan, 5-FU y leucovorina. En la tabla 7 se describen las modificaciones recomendadas de las dosis de irinotecan y cisplatino para el inicio de cada ciclo de terapia. En la tabla 8 se describen las recomendaciones para las modificaciones de las dosis durante un ciclo de terapia. Todas las modificaciones de las dosis se deben basar en la peor toxicidad precedente. No se debe iniciar un nuevo ciclo de terapia hasta que la toxicidad se haya recuperado a un grado 2 del NCI o menor. El tratamiento se puede retrasar 1 a 2 semanas para permitir que el paciente se recupere de la toxicidad relacionada con el tratamiento. Si el paciente no se ha recuperado, se debe considerar interrumpir el irinotecan.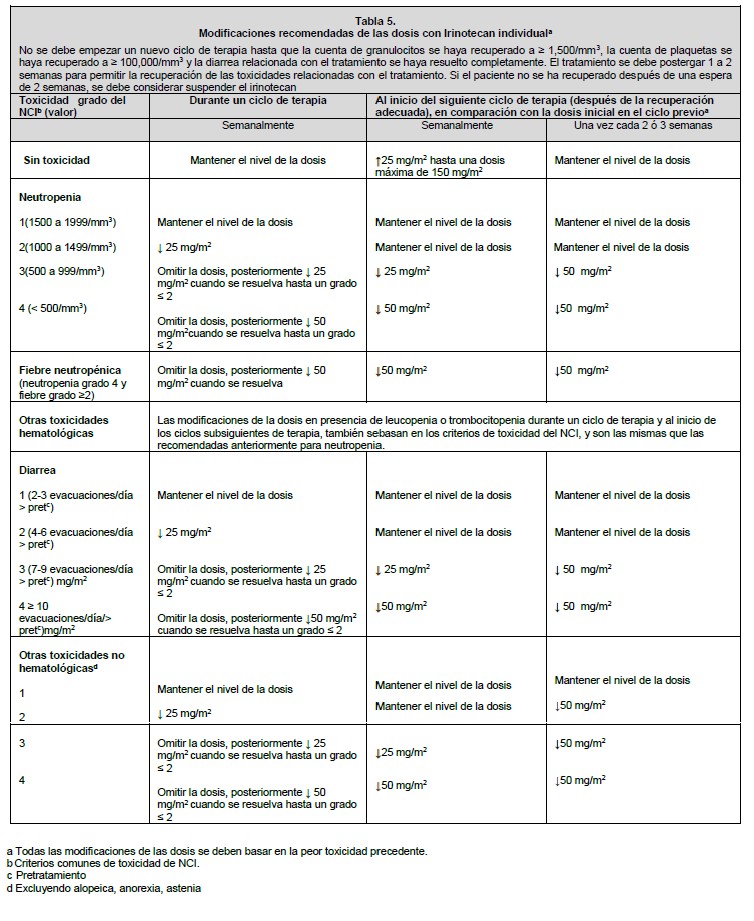
Modificaciones recomendadas de las dosis para los esquemas de combinación de irinotecan/5-fluorouracilo/leucovorina: No se debe empezar un nuevo ciclo de terapia hasta que la cuenta de granulocitos se haya recuperado a ≥ 1,500/mm3, la cuenta de plaquetas se haya recuperado a ≥ 100,000/mm3 y la diarrea relacionada con el tratamiento se haya resuelto completamente. El tratamiento se debe retrasar 1 a 2 semanas para permitir la recuperación de las toxicidades relacionadas con el tratamiento. Si el paciente no se ha recuperado después de una espera de 2 semanas, se debe considerar suspender el irinotecan.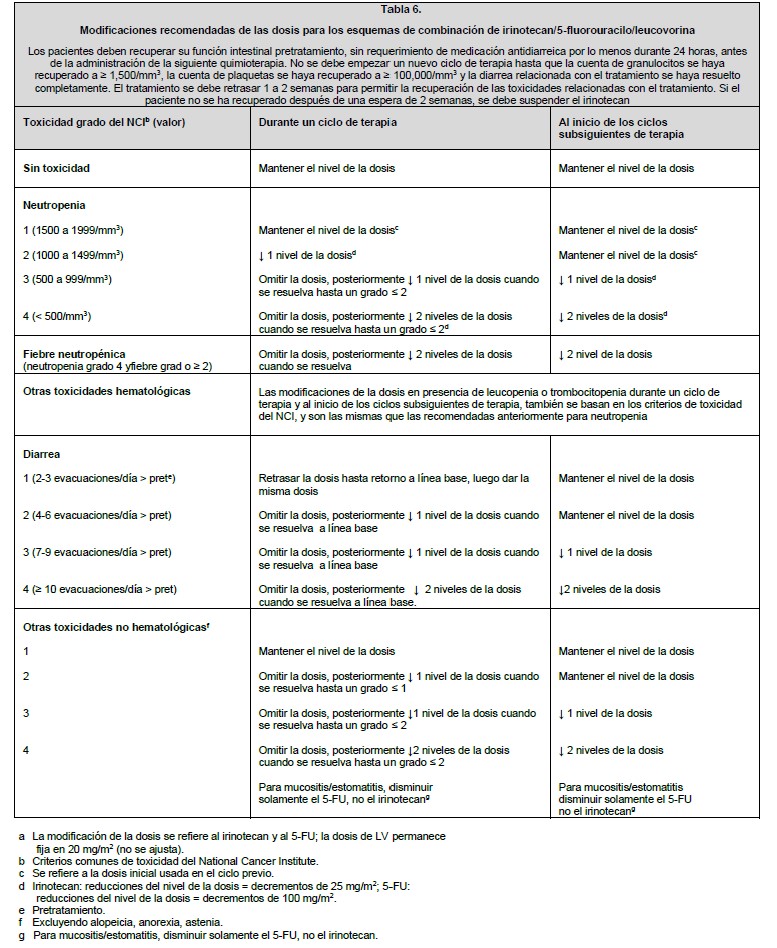
Manifestaciones y manejo de la sobredosificación o ingesta accidental: Se han administrado dosis únicas de hasta 750 mg/m2 de irinotecan en pacientes con diversos cánceres. Los eventos adversos en estos pacientes fueron similares a los reportados con las dosis y regímenes recomendados. Se debe instituir el cuidado máximo de soporte para prevenir la deshidratación debida a diarrea y tratar cualquier complicación infecciosa. No hay antídoto conocido para la sobredosificación con irinotecan.
Presentaciones: Caja con un frasco ámpula con 100 mg/5 ml. Caja con un frasco ámpula con 40 mg/2 ml.
Recomendaciones sobre almacenamiento: Consérvese en lugar fresco y seco. Las soluciones diluidas de dextrosa al 5% se deben conservar en refrigeración entre 2°C a 8°C durante 48 horas. No se congele. Consérvese a temperatura ambiente, a no más de 30°C. Protéjase de la luz. La refrigeración de las soluciones diluidas con cloruro de sodio al 0.9%, no son recomendables, debido a una baja y esporádica incidencia de partículas visibles.
Leyendas de protección: Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. No se use durante el embarazo o lactancia. No se administre si la solución no es transparente, si contiene partículas en suspensión o sedimento. No se administre si el cierre ha sido violado. Medicamento de alto riesgo. Este medicamento deberá ser administrado únicamente por médicos especialistas en oncología y con experiencia en quimioterapia antineoplásica. Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx
Nombre y domicilio del laboratorio: Hecho por: Fresenius Kabi Oncology Limited, Village Kishanpura, Baddi, Tehsil Nalagarh, Distrito Solan H.P. 174101, India. Acondicionado y Distribuido por: Fresenius Kabi México S.A. de C.V., Av. Paseo del Norte No. 5300-A, Col. San Juan de Ocotán, C.P. 45010, Zapopan, Jalisco, México.
Número del registro del medicamento: 169M2011 SSA, IV