DEPAKENE
ABBOTT
Cápsulas y jarabe
Denominación genérica: Ácido valproico.
Forma farmacéutica y formulación: Cápsulas, jarabe. Cada cápsula contiene: Ácido valproico 250 mg. Excipiente, cbp 1 cápsula. Cada 100 ml contienen: Valproato de sodio equivalente a 5.000 g de ácido valproico. Vehículo, cbp 100 ml.
Indicaciones terapéuticas: Anticonvulsivante. El ácido valproico está indicado como medicamento único o combinado en el tratamiento de la epilepsia. El ácido valproico puede ser indicado en el caso de crisis parciales, simples/complejas y/o generalizadas como son las crisis de ausencia, tónicas, clónicas y mioclónicas. El ácido valproico también se puede administrar concomitantemente en pacientes con crisis convulsivas de múltiples tipos, entre las que se incluyen crisis psicomotoras y sintomáticas.
Farmacocinética y farmacodinamia: Absorción rápida, después de la administración oral de una dosis única el nivel máximo plasmático del ácido valproico se alcanza entre una y cuatro horas después. La vida media tiene rango de 6 a 16 horas. Una vida media de rango más bajo se presenta en pacientes que toman otros antiepilépticos, o depende de la edad: la vida media del ácido valproico en niños < 10 días de edad tiene un rango de 10 a 67 horas. En niños < 2 meses de edad es de 7 a 13 horas. Los alimentos retardan ligeramente la absorción del ácido valproico pero no la absorción total. En fase de depuración del ácido valproico existe diferencia entre los niños ( > 50% ml/min/kg) > 10 años. En pacientes seniles la depuración se reduce hasta un 39% y la fracción libre de ácido valproico se incrementa en un 44%. Distribución: Se distribuye rápidamente en concentraciones terapéuticas, el 90% se une a las proteínas plasmáticas. El aumento de la dosis puede producir disminución de esta unión a las proteínas plasmáticas, lo que produce cambios en la depuración y eliminación del valproato. La eliminación del valproato y sus metabolitos es a través de la orina y muy poca por heces y vías respiratorias. El medicamento es metabolizado primero en hígado y excretado como un conjugado glucurónido. La enfermedad hepática (ver Contraindicaciones) deteriora la capacidad de eliminar el ácido valproico. La depuración de ácido valproico puede estar reducida hasta un 50%. La enfermedad renal reduce la capacidad de unión a proteínas por el ácido valproico.
Contraindicaciones: El ácido valproico no deberá administrarse a pacientes con enfermedad o disfunción hepática. Está contraindicado en pacientes con hipersensibilidad conocida al medicamento. De igual forma, está contraindicado en pacientes con trastorno en el ciclo de la urea. Deberá tenerse precaución cuando se administre el ácido valproico a pacientes con antecedentes de enfermedad hepática, pacientes con epilepsia grave acompañada de retraso mental y los que tienen enfermedad cerebral orgánica. Se han presentado alteraciones de la función hepática, incluyendo insuficiencia con resultados fatales en algunos pacientes que recibían ácido valproico. Esto ha ocurrido durante los seis primeros meses de tratamiento. La hepatotoxicidad severa o fatal puede verse precedida de síntomas inespecíficos como la pérdida del control de la epilepsia, malestar general, debilidad, letargia, edema facial, anorexia y vómito. Se deben practicar pruebas funcionales hepáticas antes de iniciar el tratamiento y a intervalos frecuentes posteriormente, en especial durante los primeros seis meses. Sin embargo, no se deberá confiar totalmente en las pruebas funcionales (bioquímica sérica), ya que éstas tal vez no sean anormales en todos los casos; por ello, deberán considerarse los resultados de un examen médico cuidadoso, efectuado periódicamente. La experiencia ha indicado que los niños menores de dos años están en mayor riesgo de desarrollar hepatotoxicidad fatal; su uso a esta edad debe hacerse con precaución extrema y como único medicamento. El beneficio del control de las crisis debe calcularse en contra del riesgo. En pacientes de mayor edad la incidencia de hepatotoxicidad disminuye. El medicamento debe descontinuarse de inmediato en presencia de insuficiencia hepática. En algunos casos la disfunción hepática ha progresado a pesar de la descontinuación del medicamento. La frecuencia de efectos adversos (particularmente elevación de las enzimas hepáticas), puede ser dependiente de las dosis. El beneficio de mejorar el control de las crisis, que se acompaña del uso de dosis mayores, debe ser sopesado en contra de la posible mayor incidencia de efectos adversos.
Precauciones generales: Hepatotoxicidad: La insuficiencia hepática total ha ocurrido en algunos pacientes en tratamiento con ácido valproico. Pancreatitis: Casos de pancreatitis se han reportado en niños y adultos que están en tratamiento con ácido valproico. Trastornos del ciclo de la urea (TCU): La encefalopatía hiperamonémica, algunas veces fatal, se ha reportado seguido al inicio del tratamiento o en pacientes con TCU. Debido a informes de trombocitopenia, inhibición de la segunda fase de agregación plaquetaria y parámetros anormales de coagulación (baja de fibrinógenos), se recomienda hacer cuentas de plaquetas y pruebas de coagulación antes de iniciar el tratamiento y a intervalos periódicos; estas pruebas también se deben hacer en pacientes que reciben algún tipo de valproato antes de planear alguna cirugía. La presencia de hemorragias, hematomas o trastornos de hemostasia/coagulación es una indicación para reducir la dosis de valproato o descontinuar el tratamiento. La ideación suicida puede ser una manifestación de trastornos psiquiátricos preexistentes, el tratamiento inicial con valproato se debe acompañar de una supervisión cercana del riesgo del paciente. Como el ácido valproico puede producir depresión del SNC, especialmente combinado con otros depresores del SNC (por ejemplo, alcohol), los pacientes deben ser aconsejados de no comprometerse a actividades peligrosas, como conducir automóviles u operar maquinaria peligrosa, hasta que se sepa que ellos no estarán adormilados con el medicamento. La seguridad y eficacia del ácido valproico para el tratamiento de la manía aguda y profilaxis de las migrañas no han sido estudiadas en individuos de 18 y 16 años, respectivamente. No hay suficiente información disponible para discernir la eficacia y seguridad de los valproatos en la profilaxis de migraña, en pacientes mayores de 65 años. Reacciones de hipersensibilidad multi-orgánica se han reportado después del inicio del ácido valproico.
Restricciones de uso durante el embarazo y la lactancia: El ácido valproico puede producir teratogenicidad, La incidencia de defectos en la médula espinal (por ejemplo, espina bífida) en el feto, puede verse aumentada en las madres que reciben el ácido valproico durante el primer trimestre del embarazo. Se ha asociado retraso en el desarrollo en el producto de mujeres embarazadas que reciben ácido valproico durante el embarazo. Cualquier medicamento antiepiléptico deberá administrarse a mujeres que pudiesen estar embarazadas solamente si se demuestra que el medicamento es esencial para el manejo de la epilepsia. Toda mujer en edad reproductiva que requieran ácido valproico, deberá ser prevenida sobre el riesgo de usarlo durante el embarazo. Lactancia: El valproato se excreta en la leche materna (1 a 10% de la concentración sérica), no se sabe qué efecto podría tener esto en el niño amamantado. Se debe tener precaución cuando se administre ácido valproico a madres en lactancia.
Reacciones secundarias y adversas: El ácido valproico generalmente se combina con otros anticonvulsivantes, así que en muchos casos no es fácil determinar si las reacciones son de ácido valproico o de la combinación. Gastrointestinales: Los efectos secundarios más comúnmente observados al comienzo del tratamiento son: náuseas, vómito e indigestión. Estos efectos generalmente son pasajeros y rara vez requieren la suspensión del tratamiento. Además, se han observado diarrea, dolores abdominales tipo cólico y constipación. También se ha informado la presencia de anorexia, pérdida de peso o aumento del apetito y peso. Efectos en el sistema nervioso central: Se han observado efectos de sedación en pacientes que reciben ácido valproico solo, pero se encuentran por lo común en pacientes que reciben tratamiento combinado. La sedación desaparece al disminuir los otros medicamentos anticonvulsivos. Son frecuentes la ataxia y cefalea, nistagmo, diplopía, "manchas en los ojos", disartria, mareo e incoordinación. Se han comunicado raros casos de coma en pacientes que también recibían fenobarbital. Dermatológicas: Aumento pasajero de la caída de pelo. Rara vez se ha observado erupción cutánea y petequias. Psiquiátricas: Se ha informado acerca de trastornos emocionales, depresión, psicosis, agresión, hiperactividad y deterioro de la conducta. Musculoesqueléticas: Se ha observado debilidad. Hepáticas: Las elevaciones discretas de las transaminasas (TGO y TGP), y deshidrogenasa láctica, son frecuentes y al parecer relacionadas con la dosis. Ocasionalmente, los cambios en las pruebas de laboratorio pueden incluir aumento de bilirrubina sérica y cambios anormales en otras pruebas de funcionamiento hepático. Estos resultados pueden reflejar hepatotoxicidad potencialmente severa. Pancreáticas: Se han presentado casos de pancreatitis aguda en niños y adultos, algunos casos de pancreatitis y hemorragia mortal. Los casos han sido poco frecuentes, pero pueden ocurrir al inicio del tratamiento o después de varios años de uso del ácido valproico o sus derivados. Hasta la fecha se han informado 431 casos de pancreatitis asociada con el uso de ácido valproico (23 casos), valproato de sodio (207 casos) y valproato semisódico (201 casos). Hematológicas: Se ha reportado trombocitopenia. El ácido valproico inhibe la segunda fase de la agregación plaquetaria; esto produce trastornos del tiempo de sangrado, petequias, formación de hematomas y hemorragias. Se ha encontrado también linfocitosis, macrocitosis e hipofibrinogenemia. También leucopenia y eosinofilia; anemia y supresión de médula ósea. Endrocrinas: Se ha reportado irregularidades en el ciclo menstrual, amenorrea secundaria, y muy rara vez la presencia de galactorrea; trastornos tiroideos anormales y pancreatitis aguda (incluyendo algunos casos de muerte). Metabólicas: Uremia, hiperglucemia, algunas veces asociada a muerte en pacientes con hiperglucemia no cetósica preexistente. Hipersensibilidad Multiorgánica Reactiva: Se ha reportado en raros casos poco después de haber iniciado el tratamiento con valproato en la población adulta y pediátrica.
Interacciones medicamentosas y de otro género: El ácido valproico tiene interacciones farmacológicas con diversos medicamentos, principalmente con otros antiepilépticos. Debido a que posee un amplio rango de indicaciones, el uso concomitante con otros fármacos es frecuente, lo que genera la posibilidad de interacciones importantes, como las siguientes: Fármacos y sustancias con interacción con ácido valproico: Alcohol: El ácido valproico puede potenciar la actividad depresora del alcohol y los barbitúricos sobre el S.N.C. Ácido acetilsalicílico, warfarina, carbamazepina y dicumarol: Se recomienda precaución cuando se administre ácido valproico a pacientes que ingieren otros medicamentos que afectan la coagulación como los anticoagulantes y el ácido acetilsalicílico. Barbitúricos: Se ha informado un aumento en los niveles séricos de fenobarbital que condicionan depresión grave del SNC. Se recomienda no utilizar el ácido valproico conjuntamente con el fenobarbital, por haberse encontrado un potencial mayor de hepatotoxicidad con esta asociación. Clonazepam: El uso concomitante de ácido valproico y clonazepam puede producir un estado de ausencia. Diazepam: El ácido valproico desplaza el diazepam de los sitios de unión a la albúmina plasmática. Difenilhidantoínas: Puede ocurrir aumento o disminución en los niveles séricos de difenilhidantoína en pacientes que toman ácido valproico. Se recomienda precaución cuando se administre concomitantemente con medicamentos que afecten la coagulación con warfarina. Felbamato: Se ha demostrado en estudios que incrementa los niveles de concentración del ácido valproico. Haloperidol: No se reporta interacciones que modifiquen los niveles plasmáticos del ácido valproico. Lamotrigina: En co-administración con ácido valproico la vida media de lamotrigina se incrementa de 26 a 70 h. Topiramato: La administración concomitante de ácido valproico y topiramato a sido asociado con hiperamonemia con y sin encefalopatía.
Alteraciones en los resultados de pruebas de laboratorio: Ha habido informes de alteraciones de las pruebas de función tiroidea asociados a ácido valproico, se desconoce su significación clínica. Como se elimina parcialmente por orina como metabolito cetónico, puede producir falsa interpretación de la prueba de cetonas.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Los estudios en animales han demostrado que el ácido valproico produce teratogenicidad. Dosis mayores de 65 mg/kg/día administradas a ratas y ratones preñadas producen anormalidades óseas, dosis mayores de 150 mg/kg/día administradas en conejas preñadas producen reabsorción fetal y anormalidades de tejidos blandos. Se ha observado en las ratas, con esa misma dosis. La suspensión de la medicación en mujeres debe ser valorada si se hace antes del embarazo o durante el mismo, ya que no podemos asegurar que inclusive convulsiones menores, produzcan problemas en el desarrollo del embrión o feto. Carcinogénesis: La administración de ácido valproico en ratas y ratones a dosis de 80 y 170 mg/kg/día durante dos años produjo un efecto carcinogénico neoplásico en ambas especies. Mutagénesis: No hay evidencia de acción mutagénica. Fertilidad: Los estudios de toxicidad crónica realizados en ratas y perros, jóvenes y adultos, con dosis mayores de 200 mg/kg/día y 90 mg/kg/día respectivamente, produjeron reducción de la espermatogénesis y atrofia testicular. Se desconoce la acción que ácido valproico pueda tener sobre el desarrollo testicular, producción espermática y fertilidad en el humano.
Dosis y vía de administración: El ácido valproico se administra por vía oral. La dosis inicial recomendada tanto en niños como en adultos es de 15 mg/kg/día hasta controlar las convulsiones o si aparecen efectos secundarios que impidan mayores aumentos. La dosis máxima recomendada es de 60 mg/kg/día dividida en varias tomas. Si el total de la dosis es mayor de 250 mg, también debe administrarse fraccionada. Dosis inicial: 15 mg/kg/día. Dosis de ajuste: 5 a 10 mg/kg/día. Dosis máxima: 60 mg/kg/día.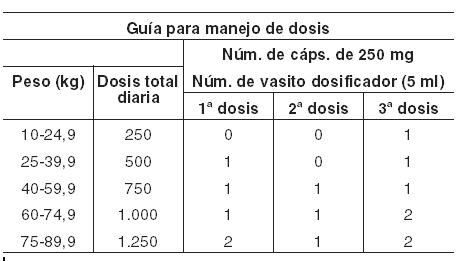
Para evitar la irritación gastrointestinal puede administrarse el medicamento con los alimentos o aumentando lentamente la dosis desde un nivel inicial bajo. Si la dosis por toma excede los 250 mg, se sugiere administrar la dosis complementaria después de la cena y después del desayuno. Las cápsulas deben ser deglutidas sin masticar, para evitar la irritación local de la boca y la garganta.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: La sobredosis con ácido valproico puede producir coma profundo, ya que el ácido valproico se absorbe rápidamente, se realizará lavado gástrico dependiendo del tiempo que haya transcurrido desde la ingesta para que sea de utilidad. Se ha reportado que la administración de naloxona revierte los efectos depresivos que el ácido valproico tiene sobre el S.N.C. Además, se ha reportado el uso de L-carnitina para revertir los efectos tóxicos del ácido valproico a nivel hepático. Debe usarse con precaución, ya que también revierte el efecto anticonvulsivante.
Presentación(es): DEPAKENE (ácido valproico) se presenta en frascos con 30 y 60 cápsulas con 250 mg de ácido valproico. DEPAKENE jarabe, frasco con 100 ml. Cada vaso dosificador graduado (5 ml) contiene 250 mg de ácido valproico (equivalente a 288 mg de valproato de sodio).
Recomendaciones sobre almacenamiento: DEPAKENE CÁPSULAS: Consérvese a temperatura ambiente a no más de 25°C, en lugar seco. DEPAKENE JARABE: Consérvese a temperatura ambiente a no más de 30°C, en lugar seco.
Leyendas de protección: Producto perteneciente al grupo IV. Literatura exclusiva para médicos. No se deje al alcance de los niños. Su venta requiere receta médica.
Nombre y domicilio del laboratorio: Abbott Laboratories de México, S.A. de C.V. Calzada de Tlalpan No. 3092, Col. Ex - Hacienda Coapa, Coyoacan, México D.F., C.P. 04980.
Número de registro del medicamento: Cápsulas: 0542M79 SSA IV. Jarabe: 0543M79 SSA IV.
Clave de IPPA: GEAR-06350122070060/RM2007. JEAR-06350122070062/RM2006.
DEPAKENE
ABBOTT
Solución inyectable
Denominación genérica: Ácido Valproico.
Forma farmacéutica y formulación: Solución Inyectable. El frasco ámpula contiene Valproato de sodio equivalente a 500 mg de ácido valproico. Agua inyectable cbp 5 ml.
Indicaciones terapéuticas: DEPAKENE está indicado en el manejo agudo de la epilepsia, tanto parcial como generalizada. Como alternativa intravenosa en pacientes en quienes la administración por vía oral, de otros productos de valproato, no es factible temporalmente, en las siguientes condiciones: DEPAKENE está indicado en la impregnación para el control de la epilepsia. Como monoterapia o tratamiento adjunto en pacientes con epilepsia. Existen reportes en el que se ha usado en los episodios agudos de migraña con resultados favorables.
Farmacocinética y farmacodinamia: DEPAKENE es la sal sódica del ácido valproico y está químicamente designado como sodio 2-propilpentanol. Su peso molecular es de 166.2. Es un polvo blanco, sin olor, cristalino. Su pH es de 7.6 y contiene únicamente 0.4 mg de acetato disódico. Farmacocinética. Biodisponibilidad. Dosis equivalentes a valproato IV y valproato oral resultan en Cmáx y Cmín equivalentes y en la exposición sistémica total al ión valproato. Sin embargo, la proporción de absorción del ión valproato puede depender de la formulación utilizada. El valproato semisódico en tabletas o el valproato de sodio para vía IV (infusión para una hora), ambas en dosis de 250 mg, cada 6 horas, durante 18 días, en voluntarios masculinos, en estado estable resultó con ABC de Cmáx, Cmín equivalentes, al igual que después de la primera dosis. El Tmáx después del valproato de sodio IV ocurre hacia el final de la hora de infusión, mientras que el Tmáx posterior a la dosis oral de valproato semisódico ocurrió a las 4 horas, aproximadamente. Debido a que la cinética del valproato no unido es lineal, se puede asumir la bioequivalencia entre valproato semisódico y valproato de sodio, a las dosis máximas recomendadas de 60 mg/kg/día. También fueron equivalentes el ABC de la Cmáx resultantes de la administración de valproato IV 500 m, como una sola infusión durante 1 hora y en una sola dosis de 500 mg de ácido valproico en jarabe a 17 adultos masculinos sanos. Los pacientes mantenidos con dosis diarias de 750 mg o 4250 mg de ácido valproico (en dosis divididas cada 6 horas) como sólo con valproato semisódico oral (n=24) o con otro fármaco antiepiléptico estabilizado (carbamazepina, n=15; fenitoína, n=11 o fenobarbital, n=1) mostraron niveles plasmáticos comparables de ácido valproico cuando se cambió del valproato semisódico oral al valproato de sodio IV (infusión de 1 hora). Distribución: El valproato de sodio existe como ión valproato en la sangre. La unión del valproato a las proteínas plasmáticas depende de la concentración y la fracción libre. En el anciano la unión a proteínas se reduce, así como en pacientes con enfermedades hepáticas crónicas, insuficiencia renal y por la presencia de otros fármacos (por ejemplo, aspirina). Por el contrario, el valproato puede desplazar ciertos fármacos unidos a las proteínas plasmáticas (por ejemplo, fenitoína, carbamazepina, warfarina y tolbutamida) (vea Precauciones, Interacciones Farmacológicas). Las concentraciones del ión valproato en el líquido cerebroespinal (LCR) son cercanos a las concentraciones libres en plasma (cerca del 10% de la concentración total). Metabolismo: El ión valproato se metaboliza casi totalmente en el hígado. En pacientes adultos, bajo monoterapia, 30% a 50% de una dosis administrada aparece en la orina como glucurónido. Otra vía metabólica secundaria pero importante, es b-oxidación mitocondrial, hasta 40% de la dosis. En general menos de 15 a 20% de la dosis es eliminada por otros mecanismos oxidativos y menos de 3% se elimina como tal, por la orina. La relación entre la dosis y la concentración total de ión valproato, es no lineal; la concentración aumenta proporcionalmente con la dosis, en vez de ello, incrementa en menor extensión debido a saturación de la unión a las proteínas plasmáticas. La cinética del fármaco no unido (libre) es lineal. Eliminación: El aclaramiento plasmático promedio y el volumen de distribución para el total del ión valproato son 0.56 l/h/173 m2 y 11 l/1.73 m2, respectivamente. La vida media terminal para el ión valproato en monoterapia después de 60 minutos de infusión intravenosa de 1000 mg, fue de 16 ± 3.0 horas. Los datos presentados se pueden aplicar principalmente a los pacientes que no están tomando fármacos que afecten los sistemas enzimáticos del hígado. Por ejemplo, los pacientes que toman antiepilépticos inductores de enzimas (carbamazepina, fenitoína, fenobarbital) pueden aclarar el valproato más rápidamente. Debido a esos cambios en el aclaramiento del ión valproato, se debe intensificar el monitoreo de las concentraciones de los antiepilépticos, siempre que se introduzca o se retire un antiepiléptico concomitante. Poblaciones especiales: Neonatos: Los menores de dos meses de edad tienen una habilidad muy reducida para eliminar el ión valproato, respecto a niños mayores y a los adultos. Esta es una consecuencia de aclaramiento reducido (debido a retraso en el desarrollo de glucuronil transferasa y otros sistemas enzimáticos implicados en la eliminación del valproato) y de aumento en el volumen de distribución (en parte por disminución de unión a proteínas plasmáticas). Por ejemplo, en un estudio, la vida media en niños menores de 10 días de edad fue de 10 a 67 horas, comparada con 7 a 13 horas en niños mayores de dos meses. Niños: Los pacientes pediátricos (por ejemplo, entre tres meses y 10 años) tienen 50% más alto aclaramiento, expresados por peso (es decir, ml/min/kg), que los adultos. Después de los 10 años, los niños tienen parámetros farmacocinéticos que se aproximan a los de los adultos. Ancianos: La capacidad de los ancianos (entre 68 a 89 años) para eliminar el ión valproato está reducida respecto a los adultos jóvenes (edad: 22 a 26 años). El aclaramiento intrínseco se reduce por 39%; la fracción libre está reducida por 44%. Por tanto, la dosis inicial debe reducirse en el anciano (vea Dosis y Administración). Género. No hay diferencias en el aclaramiento del fármaco libre (no unido) ajustado por área de superficie corporal, entre hombres y mujeres (4.8 ± 0.17 y 4.7 ± 0.07 l/h/1.73 m2, respectivamente. Raza: No se han estudiado los efectos de la raza sobre la cinética del ión valproato. Enfermedad hepática: Vea Contraindicaciones y Advertencias. La enfermedad hepática impide la capacidad de eliminar el ión valproato. En un estudio, el aclaramiento del ión valproato libre se redujo en 50% en 7 pacientes con cirrosis y en 16% en 4 pacientes con hepatitis aguda, comparados con 6 sujetos sanos. En tal estudio, la vida media del ión valproato aumentó de 12 a 18 horas. La enfermedad hepática se asocia con disminución de las concentraciones de albúmina y de mayores fracciones de ión valproato libre (no unido) (2 a 2.6 veces mayor). Por ello, el monitoreo de las concentraciones totales puede ser equivocado, puesto que las concentraciones libres pueden estar sustancialmente elevadas en pacientes con enfermedad hepática mientras que las concentraciones totales pueden parecer normales. Enfermedad renal: Se ha informado de una ligera reducción (27%) de la eliminación del ión valproato no unido, en pacientes con insuficiencia renal (aclaramiento de creatinina < 10 ml/min); sin embargo, la hemodiálisis típicamente reduce las concentraciones del ión valproato cerca de 20%. Por tanto, parece que no es necesario ajustar la dosis en pacientes con insuficiencia renal. En estos pacientes, la unión a proteínas está sustancialmente reducida; por ello, el monitoreo de las concentraciones totales puede llevar a errores. Niveles plasmáticos y efecto clínico: Las relaciones entre concentraciones plasmáticas y respuesta clínica no están bien documentadas. Un factor contribuidor es la concentración no lineal, dependiente de la unión a proteínas, del ión valproato, lo cual afecta el aclaramiento del fármaco. Así, el monitoreo del ión valproato sérico total no proporciona un índice confiable de la fracción activa del ión valproato. Por ejemplo, debido a que la unión del ión valproato a las proteínas plasmáticas es dependiente de la concentración, la fracción libre aumenta desde cerca de 10% a 40 mcg/ml hasta 18.5% al llegar a 130 mcg/ml, de concentración. Ocurren fracciones libres mayores a las esperadas en los ancianos, en los pacientes hiperlipidémicos y en aquellos con insuficiencia renal y hepática. Epilepsia: En la epilepsia, el rango terapéutico comúnmente se considera entre 50 a 100 mcg/ml del ión valproato total, si bien algunos pacientes pueden ser controlados con menores o mayores concentraciones plasmáticas que las referidas. Las dosis equivalentes del valproato sodio IV y valproato semisódico producen niveles plasmáticos equivalentes del ión valproato (vea Farmacología Clínica, Farmacocinética). Los estudios clínicos con valproato semisódico por vía oral en pacientes epilépticos con crisis parciales complejas (CPC), han mostrado reducir la incidencia de las CPC, ya sean solos o asociados con otros antiepilépticos.
Contraindicaciones: Valproato de sodio IV, no debe ser administrado a pacientes con enfermedad hepática o insuficiencia hepática significativa. Ni en sujetos con hipersensibilidad conocida al valproato de sodio. Advertencias: Ha ocurrido la muerte en sujetos con insuficiencia hepática que recibieron ácido valproico; usualmente durante los primeros seis meses de tratamiento, precedidas por síntomas inespecíficos: malestar, debilidad, letargia, edema facial, anorexia y vómito; perdida del control de convulsiones. Se requiere control estrecho mediante pruebas de función hepática (PFH), antes de usar valproato y luego a intervalos frecuentes, principalmente los seis primeros meses. No se debe confiar sólo en PFH ya que pueden no ser normales, y basarse en datos clínicos y físicos. Precaución especial en caso de antecedentes de insuficiencia o enfermedad hepática: Uso de varios anticonvulsivantes, niños, pacientes con trastornos metabólicos congénitos, convulsiones severas y retraso mental, enfermedad cerebral orgánica. Los menores de 2 años de edad tienen mayor riesgo de hepatotoxicidad fatal.
Precauciones generales: Insuficiencia hepática (ver Contraindicaciones): Se debe tener precaución al administrar productos con valproato en personas con antecedentes de enfermedades hepáticas. Pancreatitis: Algunos casos de pancreatitis se han reportado en niños y adultos que estaban usando productos de valproato. Se han descrito casos hemorrágicos de rápida evolución. Deben los pacientes ser advertidos sobre síntomas como dolor abdominal, náusea, vómito y/o anorexia, ya que pueden ser síntomas iniciales de pancreatitis. Hiperamonemia: La hiperamonemia ha sido reportada en casos asociados a tratamiento con cualquier tipo de valproato y puede presentarse aún en pacientes con examen de función hepática normal. Encefalopatía hiperamonemica asociada a la administración concomitante de topiramato. La administración concomitante de topiramato al tratamiento con cualquier valproato está asociada a hiperamonemia con o sin encefalopatía. Los datos clínicos incluyen alteración aguda del estado de conciencia, vómito y letargo. Debido a informes de trombocitopenia, inhibición de la segunda fase de agregación plaquetaria y parámetros anormales de coagulación (baja de fibrinógenos), se recomienda hacer cuentas de plaquetas y pruebas de coagulación antes de iniciar el tratamiento y a intervalos periódicos; estas pruebas también se deben hacer en pacientes que reciben algún tipo de valproato antes de planear alguna cirugía. La presencia de hemorragias, hematomas o trastornos de la hemostasia/coagulación es una indicación para reducir la dosis de valproato o descontinuar el tratamiento. Como el valproato de sodio IV puede producir depresión del SNC, especialmente combinado con otros depresores del SNC (por ejemplo, alcohol), los pacientes deben ser aconsejados de no comprometerse a actividades peligrosas, como conducir automóviles u operar maquinaria peligrosa, hasta que se sepa que ellos no estarán adormilados con el medicamento. No existe suficiente información para discernir la eficacia y seguridad de los valproatos en la profilaxis de migraña, en pacientes mayores de 65 años.
Restricciones de uso durante el embarazo y la lactancia: El ácido valproico puede producir teratogenicidad. La incidencia de defectos en la médula espinal (por ejemplo, espina bífida) en el feto, puede verse aumentada en las madres que reciben el ácido valproico durante el primer trimestre del embarazo. Se ha asociado retraso en el desarrollo del producto de mujeres embarazadas que reciben ácido valproico. Cualquier medicamento antiepiléptico deberá administrarse a mujeres que pudiesen estar embarazadas solamente si se demuestra que el medicamento es esencial para el manejo de la epilepsia. Toda mujer en edad reproductiva que requieran ácido valproico, deberá ser prevenida sobre el riesgo de usarlo durante el embarazo. Lactancia: El valproato se excreta en la leche materna (1 a 10% de la concentración sérica), no se sabe que efecto podría tener esto en el niño amamantado. Se debe tener precaución cuando se administre ácido valproico a madres en lactancia. Advertencias: Debido a informes de trombocitopenia, inhibición de la fase secundaria de la agregación plaquetaria y parámetros de coagulación anormales (Ej.: bajo fibrinógeno) se recomienda realizar la cuenta de plaquetas y pruebas de coagulación antes de iniciar el tratamiento y a intervalos periódicos después, así como antes de cualquier cirugía electiva. La evidencia de hemorragia, magulladuras o un trastorno de hemostasia/coagulación, es un indicación para la reducción de la dosis de valproato de sodio IV o para suspender su uso. Se ha informado de hiperamonemia, con o sin letargia o coma, en ausencia de pruebas de función hepática anormales, con más frecuencia que la hiperamonemia sintomática, que demanda mayor frecuencia de monitoreo; si ocurren síntomas clínicos significativos, el tratamiento con valproato de sodio IV debe ser modificado o de plano suspendido. Ya que el valproato de sodio IV puede interactuar con fármacos inductores enzimáticos, administrados concurrentemente, se deben determinar periódicamente las concentraciones plasmáticas de ambos fármacos durante el inicio del tratamiento conjunto (vea Precauciones e Interacción de Fármacos). Los niños menores de dos años de edad tienen un riesgo considerablemente mayor de desarrollar hepatotoxicidad fatal. No se ha determinado la seguridad del valproato de sodio inyectable en niños de esta edad. Por tanto, si se toma la decisión de utilizarlo en ellos, deberá hacerlo con extrema precaución y como único agente. Se deben sopesar los beneficios de la inyección contra los riesgos. Por arriba de dos años de edad, la experiencia en la epilepsia ha señalado que la incidencia de hepatotoxicidad fatal disminuye considerablemente en grupos de pacientes progresivamente de mayor edad. Los niños, en especial aquellos que reciben fármacos inductores enzimáticos, requerirán mayores dosis de mantenimiento para obtener las concentraciones de ácido valproico totales y libres deseadas. La variabilidad en los límites de la fracción libre limita la utilidad clínica del monitoreo de la concentración plasmática total de ácido valproico. La interpretación de las concentraciones de ácido valproico en niños debe considerar los factores que afectan el metabolismo hepático y la unión a proteínas plasmáticas. No se identificaron aspectos importantes respecto a la seguridad en ensayos clínicos con 24 pacientes entre 2 a 17 años de edad, que recibieron valproato de sodio inyectable, ni en 19 pacientes > 65 años de edad. La toxicología básica y las manifestaciones patológicas del valproato de sodio en ratas recién nacidas y jóvenes (4 y 14 días de edad, respectivamente) son similares a las observadas en ratas adultas jóvenes. Sin embargo, adicionalmente se han encontrado alteraciones renales en las jóvenes y alteraciones renales con displasia retinal en las recién nacidas, con dosis de 240 mg/kg/día (equivalente a la dosis máxima diaria humana con base en mg/m2). Tales anomalías no se observaron con 90 mg/kg/día (40% de la dosis humana con mg/m2).
Reacciones secundarias y adversas: Los eventos adversos que induce la inyección de valproato de sodio incluyen a los observados con formas orales del mismo. La forma inyectable, por lo general, ha sido bien tolerada en ensayos clínicos realizados en voluntarios adultos masculinos y en pacientes epilépticos, en dosis de 125 a 6000 mg (dosis total / día). Un total de 2% de pacientes discontinuaron el tratamiento con valproato de sodio inyectable debido a eventos adversos, siendo los más comunes (2 casos cada uno) náusea / vómito y amilasa elevada. Otros, que llevaron a la suspensión del uso del fármaco fueron: alucinaciones, neumonía, cefalea, reacción en el sitio de inyección y marcha anormal. El mareo y el dolor en el sitio de inyección fueron observados con mayor frecuencia con una velocidad de infusión de 100 mg/min que a velocidades de hasta 33 mg/min. A velocidad de 200 mg/min, el mareo y la perversión del sabor ocurrieron con mayor frecuencia que a 100 mg/min. La velocidad máxima de infusión estudiada, fue de 200 mg/min. Los eventos adversos informados por al menos 0.5% de todos los sujetos / pacientes en los ensayos clínicos con valproato de sodio inyectable se muestran, en resumen, en la Tabla 3.
Epilepsia: Crisis Parciales Complejas. Con base en ensayos controlados con placebo, del tratamiento adjunto para el tratamiento de las crisis parciales complejas (CPC), por lo general, el divalproato de sodio fue bien tolerado, con la mayoría de eventos adversos calificados de severidad leve a moderada. La intolerancia fue la principal razón para la discontinuación en los pacientes tratados con divalproato de sodio (6%) comparado con los pacientes tratados con placebo (1%).
En la Tabla 3 se enlistan eventos adversos que surgen durante el tratamiento y que fueron informados por ≥ 5% de pacientes en el grupo con la dosis más alta de divalproato de sodio y en quienes la incidencia fue mayor para el grupo con la dosis más baja, en ensayos como monoterapia de convulsiones parciales complejas. Puesto que, durante la primera parte del ensayo, los pacientes habían sido titulados en retirada de otro fármaco antiepiléptico, no es posible en muchos casos determinar en qué casos los siguientes eventos adversos pueden ser atribuidos al divalproato de sodio solo, o a la combinación del valproato semisódico y otros fármacos antiepilépticos.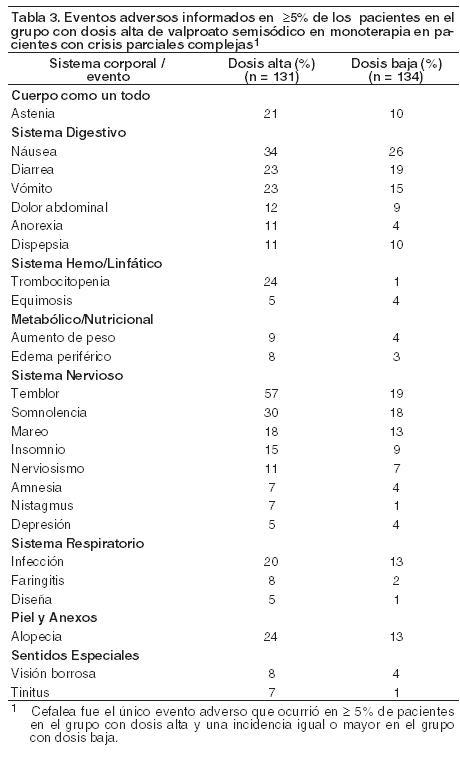
Los siguientes eventos adversos adicionales fueron informados en proporción > 1% y < 5% en 358 pacientes tratados con divalproato de sodio, en los ensayos controlados, por convulsiones parciales complejas. Cuerpo total: Dolor de espalda, dolor torácico y malestar; cardiovascular: taquicardia, hipertensión y palpitaciones; digestivo: hiperfagia, flatulencia, hematemesis, eructos, pancreatitis y abscesos periodontales; hemo/linfático: petequias; metabólico/nutricional: aumento de TGO y TGP; músculo-esqueléticos: mialgia, contracciones, artralgias, calambres en las piernas y miastenia; nervioso: ansiedad, confusión, trastornos del lenguaje, marcha anormal, parestesias, hipertonía, incoordinación, sueños anormales y trastornos de personalidad; respiratorio: sinusitis, aumento de tos, neumonía y epistaxis; piel y anexos: salpullido, prurito y piel seca; sentidos especiales: perversión del sabor, visión borrosa, trastornos en la audición, sordera y otitis media; urogenital: incontinencia urinaria, dismenorrea, amenorrea y frecuencia urinaria. Otra población de pacientes. Eventos adversos de otros estudios con epilepsia, informes espontáneos y otras fuentes: Gastrointestinal: Al iniciar la terapia, náusea, vómito e indigestión, son transitorios y rara vez se requiere suspender el tratamiento. Diarrea, calambres abdominales, constipación, anorexia con algo de pérdida de peso, hiporexia con aumento de peso. El uso de una forma de liberación retardada oral, de divalproato de sodio, puede reducir los efectos gastrointestinales. SNC: Efectos sedantes, más comunes por tratamiento de combinación, que se abate al reducir los otros antiepilépticos. Temblor (dosis-respuesta), alucinaciones, ataxia, cefalea, nistagmus, diplopía, asterixis, "manchas ante los ojos", disartria, mareo, confusión, hipoestesia, vértigo e incoordinación. Coma, en casos raros, solo o en combinación con fenobarbital. Encefalopatía, con o sin fiebre; hiperamonemia, sin evidencia de disfunción hepática o niveles plasmáticos inapropiados. La mayoría de los pacientes se recuperaron con mejoría notable de los síntomas, al discontinuar el fármaco. Atrofia cerebral reversible y demencia. Se ha reportado estados de hiperamonemia y encefalopatía asociada al uso concomitante con topiramato (Human Psychopharmacology, Clin Exp 2004;19:193-203). Dermatológicos: Caída de pelo transitoria. Salpullido cutáneo, fotosensibilidad, prurito generalizado, eritema multiforme, síndrome de Stevens-Johnson y necrólisis tóxica epidérmica (rara vez). Psiquiátricos: Alteración emocional, depresión, psicosis, agresión, hiperactividad, hostilidad y deterioro conductual. Músculo-esquelético: debilidad. Hematológico: trombocitopenia y alteración del tiempo de sangrado, petequias, magulladuras, hematoma, epistaxis y hemorragia franca (vea Precauciones); linfocitosis relativa, macrolitosis, hipofibrinogenemia, leucopenia, eosinofilia, anemia, incluyendo a la macrocítica con o sin deficiencia de fosfatos, supresión de médula ósea, pancitopenia, anemia aplástica, porfiria aguda intermitente. Hepático: Elevación menor de TGO y TGP y LDH, son frecuentes y dependen de la dosis-respuesta. Endocrino: Menstruación irregular, amenorrea secundaria, cr