EDARBI*
TAKEDA
Denominación genérica: Azilsartán medoxomilo.
Forma farmacéutica y formulación: Cada tableta contiene: Azilsartán medoxomilo de potasio 42.680 u 85.360 mg. Equivalente a: Azilsartán medoxomilo 40 u 80 mg. Excipiente cbp 1 tableta.
Indicaciones terapéuticas: EDARBI® (azilsartán medoxomilo) es un profármaco administrado de forma oral que es convertido rápidamente por las estearasas en el tracto gastrointestinal y/o durante la absorción a la fracción activa, azilsartán, un antagonista o bloqueador selectivo del receptor de angiotensina II tipo 1 (ARB) indicado para el tratamiento de la hipertensión leve a moderada. Puede ser usado solo o en combinación con otros agentes antihipertensivos con excepción de productos que contengan aliskireno o inhibidores de la enzima convertidora de angiotensina.
Contraindicaciones: Hipersensibilidad a cualquiera de los componentes de la formula. Embarazo. No administrar productos que contienen aliskireno con azilsartán medoxomilo en pacientes con diabetes. No administrar Edarbi® con inhibidores de la enzima convertidora de angiotensina.
Precauciones generales: Morbilidad y Mortalidad Fetal / Neonatal. Cuando se detecte el embarazo, EDARBI® deberá interrumpirse tan pronto como sea posible. El uso de fármacos que actúan sobre el sistema renina - angiotensina durante el segundo y tercer trimestres del embarazo reduce la función renal fetal e incrementa la morbilidad y mortalidad fetal y neonatal. Los oligohidramnios resultantes pueden ser asociados con hipoplasia pulmonar fetal y con deformaciones esqueléticas. Los potenciales efectos adversos en los neonatos incluyen hipoplasia craneal, anuria, hipotensión, falla renal y muerte. Cuando se detecte el embarazo, discontinuar la administración de EDARBI® tan pronto como sea posible. Estos efectos adversos parecen estar asociados con el uso de estos medicamentos en el segundo y tercer trimestre del embarazo. La mayoría de los estudios epidemiológicos que examinan las anormalidades fetales después de la exposición al uso de antihipertensivos en el primer trimestre no han distinguido a los fármacos que afectan el sistema renina-angiotensina de otros agentes antihipertensivos. Es importante el adecuado manejo de la hipertensión materna durante el embarazo a fin de optimizar los resultados tanto en la madre como en el feto. En el caso inusual de que no exista una alternativa apropiada de terapia con fármacos que actúen sobre el sistema renina - angiotensina para un paciente en particular, la madre deberá ser informada de los peligros potenciales para el feto y se deberán realizar exámenes seriales de ultrasonido para evaluar el ambiente intra-amniótico. Si se observa oligohidramnios, EDARBI® deberá interrumpirse a menos que se considere que salva la vida a la madre. Pueden ser realizadas pruebas fetales apropiadas, basándose en el número de semanas de embarazo. Sin embargo, las pacientes y médicos deberán estar conscientes de que los oligohidramnios pueden no aparecer hasta que el feto tenga un daño sostenido irreversible. Los infantes con historiales de exposición en el útero a EDARBI® deberán ser observados de cerca en cuanto a hipotensión, oliguria e hiperkalemia. Hipotensión en Pacientes Disminuidos de Volumen circulante o depleción salina: Puede presentarse hipotensión en pacientes hipertensos con hipovolemia y/o hiponatremia. En pacientes con un sistema renina - angiotensina activado, como pacientes disminuidos de volumen o sal (por ejemplo aquellos que son tratados con dosis altas de diuréticos), puede ocurrir hipotensión sintomática después del inicio del tratamiento con EDARBI®. Por lo tanto, la depleción salina y/o la disminución de volumen debe corregirse antes de la administración de EDARBI®, y empezar el tratamiento bajo supervisión médica cercana. Si ocurre hipotensión, el paciente deberá ser colocado en posición supina y, de ser necesario, administrarle una infusión intravenosa de solución salina. Una respuesta hipotensiva transitoria no es una contraindicación al tratamiento adicional, el cual generalmente puede ser continuado sin dificultad una vez que la presión sanguínea se haya estabilizado. Función Renal Debilitada: Monitorear el deterioro de la función renal en pacientes con insuficiencia renal. Como consecuencia de la inhibición del sistema renina - angiotensina, se pueden anticipar cambios en la función renal en individuos susceptibles tratados con EDARBI®. En pacientes cuya función renal pueda depender de la actividad del sistema renina - angiotensina (por ejemplo, pacientes con insuficiencia cardiaca congestiva severa, estenosis de arteria renal o disminución de volumen), el tratamiento con inhibidores de la enzima convertidora de angiotensina y con bloqueadores del receptor de angiotensina II, se ha asociado con oliguria y/o azoemia progresiva y raras veces con insuficiencia renal aguda y/o muerte. Se pueden anticipar resultados similares en pacientes tratados con EDARBI®. En estudios con inhibidores ACE en pacientes con estenosis de arteria renal unilateral o bilateral, se han reportado incrementos en la creatinina sérica o nitrógeno de urea en la sangre. No ha habido uso a largo plazo de EDARBI® en pacientes con estenosis de arteria renal unilateral o bilateral, pero se pueden esperar resultados similares a los previamente reportados. Estenosis de la arteria renal. La función renal puede empeorar en pacientes con estenosis de la arteria renal. Bloqueo dual sistema renina-angiotensina-aldosterona: Existe evidencia de que el uso concomitante de inhibidores de la enzima convertidora de angiotensina, bloqueadores del receptor de angiotensina II o de productos que contienen aliskireno, incrementa el riesgo de hipotensión, hipercalemia, y disminución de la función renal (incluyendo falla renal aguda). No se recomienda el bloqueo dual del sistema renina angiotensina aldosterona a través del uso combinado de inhibidores de la enzima convertidora de angiotensina, bloqueadores del receptor de angiotensina II o de productos que contienen aliskireno
Restricciones de uso durante el embarazo y la lactancia: Embarazo. No hay experiencia clínica con el uso de EDARBI® en mujeres embarazadas. Los medicamentos que actúan directamente sobre el sistema renina-angiotensina pueden causar morbilidad o muerte fetal o neonatal cuando son administrados a mujeres embarazadas durante el segundo y tercer trimestre del embarazo, por lo tanto EDARBI® no debe ser administrado durante el embarazo y debe ser descontinuado una vez que se ha detectado el embarazo en la paciente. Madres en periodo de lactancia: Se desconoce si EDARBI® se excreta en la leche humana, pero azilsartán se excreta en bajas concentraciones en la leche de ratas lactantes. Debido al potencial de efectos adversos en el infante lactante, se deberá tomar una decisión de si se interrumpe la lactancia o se interrumpe el fármaco, tomando en cuenta la importancia del fármaco para la madre.
Reacciones secundarias y adversas: Experiencia de Estudios Clínicos: Debido a que los estudios clínicos se realizan bajo condiciones muy variantes, las tasas de reacciones adversas observadas en los estudios clínicos de un fármaco no pueden ser comparadas directamente con las tasas en los estudios clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica. En estudios clínicos de Fase III, un total de 4814 pacientes fueron evaluados en cuanto a seguridad cuando fueron tratados con EDARBI® a dosis de 20, 40 u 80 mg. Para los pacientes tratados con EDARBI® de 20, 40 u 80 mg, 1704 pacientes fueron tratados por al menos 6 meses, y de éstos, 588 fueron tratados por al menos 1 año. El tratamiento con EDARBI® fue bien tolerado con una incidencia total de reacciones adversas similar a placebo. Las reacciones adversas fueron generalmente leves y no dependieron de la dosis. EDARBI® fue bien tolerado independientemente del género, edad o raza. La tasa de abandonos debidos a eventos adversos en estudios controlados con placebo fue de 2.4% (19/801) para placebo, 2.2% (24/1072) para EDARBI® 40 mg, y 2.7% (29/1074) para EDARBI® 80 mg. El evento adverso más común que lleva a la interrupción, hipotensión/ hipotensión ortostática, fue reportado en 0.4% (8/2146) pacientes aleatorizados con EDARBI® 40 mg u 80 mg comparado con un 0% (0/801) de pacientes aleatorizados con placebo. Generalmente las reacciones adversas fueron leves, no relacionadas con la dosis y de forma independiente a la edad, género y raza. En estudios controlados con monoterapia de placebo, la diarrea se reportó en 2% de los pacientes tratados con EDARBI® 80 mg al día comparándose con 0.5% en pacientes con placebo. Otras reacciones adversas con relación al tratamiento que se han reportado con una incidencia ≥0.3% y mayor que en placebo en más de 3300 pacientes tratados con EDARBI® en estudios controlados se enumeran a continuación: Trastornos Gastrointestinales: náusea. Trastornos Generales y Afecciones en el Sitio de Administración: astenia, fatiga. Trastornos Músculo esqueléticos y del Tejido Conectivo: espasmo muscular. Trastornos del Sistema Nervioso: mareo, mareo por postura. Trastornos Respiratorios, Torácicos y Mediastinales: Tos. Reacciones adversas reportadas con una incidencia del < 0.1%: Angiodema, Prurito. Reacciones adversas reportadas con una incidencia del 0.2%: Salpullido (Rash). Otras reaccciones adversas reportadas durante los estudios clínicos: Aumento en creatinina sérica. Experiencia posterior a la comercialización: Se han reportado los siguientes efectos secundarios: náusea, espasmos musculares, salpullido, prurito y angioedema. Creatinina sérica elevada: Se han visto incrementos pequeños reversibles de creatinina sérica en pacientes que recibieron 80 mg de Azilsartán medoxomilo. El incremento puede ser mayor cuando se administra con clortalidona o hidroclorotiazida. Adicionalmente, pacientes con insuficiencia renal moderada a severa antes de iniciar el tratamiento con Azilsartán medoxomilo o quienes tenían más de 75 años fueron más propensos a reportar incrementos de creatinina sérica.
Interacciones medicamentosas y de otro género: No se han observado interacciones farmacológicas clínicamente significativas en estudios de azilsartán medoxomilo o azilsartán administrado con amlodipino, antiácidos, clortalidona, digoxina, fluxonazol, gliburida, ketoconazol, metformina, pioglitazona y warfarina. Por lo tanto, EDARBI® puede ser usado de manera concomitante con estos medicamentos. En pacientes con diabetes mellitus que estén bajo tratamiento médico con EDARBI® no deberán ser tratados de manera conjunta con productos que contengan aliskireno. Asimismo se debe interrumpir el tratamiento con medicamentos que contengan aliskireno antes de iniciar el tratamiento con EDARBI®. Se debe evitar el uso de productos que contienen aliskireno con azilsartán medoxomilo en pacientes con insuficiencia renal (tasa de filtración glomerular < 60 mL/min/1.73 m2). Bloqueo simultáneo del Sistema Renina-Angiotenina-Aldosterona. El bloqueo simultáneo del Sistema Renina Angiotensina con inhibidores de la ECA, bloqueadores de los receptores de la angiotensina II o aliskireno se han asociado a un aumento en el riesgo de hipotensión, hipokalemia y cambios en la función renal (incluyendo insuficiencia renal aguda). Se debe vigilar la presión arterial, la función renal y los electrolitos en pacientes en tratamiento con azilsartán medoxomilo y otros agentes que afecten al Sistema Renina-Angiotenina-Aldosterona. No se recomienda el bloqueo simultáneo del Sistema Renina Angiotensina mediante el uso combinado de inhibidores de la ECA, bloqueadores de los receptores de la angiotensina II o aliskireno. Antiinflamatorios no esteroideos incluyendo a los Inhibidores Selectivos de la Ciclooxigenasa - 2 (Inhibidores de la COX-2). En pacientes de edad avanzada, que tengan disminución en el volumen circulante (incluyendo aquellos en terapia con diuréticos), o aquellos quienes tienen comprometida la función renal, la administración conjunta de antiinflamatorios no esteroideos (AINEs), incluyendo los inhibidores selectivos de la COX-2, con antagonistas de los receptores de la Angiotensina II, incluyendo azilsartán medoxomilo, puede resultar en un deterioro de la función renal, incluyendo una posible falla renal aguda. Estos efectos son generalmente reversibles. Se debe monitorear periódicamente la función renal en pacientes en tratamiento con azilsartán medoxomilo y terapia con antiinflamatorios no esteroideos, incluyendo a los inhibidores selectivos de la COX-2. El efecto antihipertensivo de los antagonistas del receptor de angiotensina II, incluyendo al azilsartán medoxomilo, puede ser atenuado con los antiinflamatorios no esteroideos, incluyendo a los inhibidores selectivos de la COX-2. Litio: Se han reportado incrementos reversibles de concentraciones séricas de litio así como toxicidad durante el uso concurrente de inhibidores de la ECA. Puede ocurrir un efecto similar con antagonistas del receptor de angiotensina II.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Carcinogénesis: Azilsartán medoxomilo no fue carcinogénico cuando se evaluó en estudios en ratones transgénicos (Tg.rasH2) de 26 semanas y ratas de 2 años. Las dosis más altas probadas (450 mg de azilsartán medoxomilo/kg/día en el ratón y 600 mg de azilsartán medoxomilo/kg/día en la rata produjeron exposiciones a azilsartán que son 12 (ratones) y 27 (ratas) veces la exposición promedio a azilsartán en humanos dando la dosis máxima recomendada en humanos (MRHD, 80 mg de azilsartán medoxomilo/día). M-II no fue carcinogénico cuando se evaluó en estudios en ratones Tg.rasH2 de 26 semanas y ratas de 2 años. Las dosis más altas probadas (aproximadamente 8000 mg M-II/kg/día (machos) y 11,000 mg M-II/kg/día (hembras) en el ratón y 1000 mg M-II/kg/día (machos) y hasta 3000 mg M-II/kg/día (hembras) en la rata) produjeron exposiciones que son, en promedio cerca de 30 (ratones) y 7 (ratas) veces el promedio de exposición a M-II en humanos a la MRHD. Mutagénesis: Azilsartán medoxomilo, azilsartán y M-II estuvieron desprovistos de potencial genotóxico en el ensayo de mutación inversa de Ames con Salmonella typhimurium y Escherichia coli, el ensayo in vitro de aberración cromosómica en célula de ovario de hámster chino, la prueba in vitro de mutación de gen en linfoma (tk) de ratón, la prueba ex vivo de síntesis de ADN no programado. Y el ensayo in vivo de micronúcleo de médula ósea de ratón y/o rata. Deterioro de la Fertilidad: No hubo efecto de azilsartán medoxomilo sobre la fertilidad de ratas macho o hembra a las dosis orales de hasta 1000 mg de azilsartán medoxomilo/kg/día [6000 mg/m2 (aproximadamente 122 veces la MRHD de 80 mg de azilsartán medoxomilo/60 kg en una base mg/m2]. La fertilidad de las ratas también se vio sin afectación a dosis de hasta 3000 mg M-II/kg/día. Toxicología y/o Farmacología Animal: Toxicología Reproductiva: estudios de toxicidad reproductiva indicaron que azilsartán medoxomilo no fue teratogénico cuando se administró a dosis orales de hasta 1000 mg de azilsartán medoxomilo /kg/día a ratas preñadas (122 veces la MRHD en una base mg/m2) o hasta 50 mg de azilsartán medoxomilo/kg/día a conejas preñadas (12 veces la MRHD en una base mg/m2). M-II tampoco fue teratogénica en ratas o conejos a dosis de hasta 3000 mg de M-II/kg/día. En estudios de desarrollo peri y post natal en rata, se observaron efectos adversos en la viabilidad de las crías por la administración de azilsartán medoxomilo (1.2 veces la MRHD en una base mg/m2) a ratas preñadas y en lactancia. Estudios clínicos: Los efectos antihipertensivos de EDARBI® se han demostrado en 7 estudios doble ciego, aleatorizados, controlados con placebo y controlados con comparador activo, que variaron entre 6 semanas y 6 meses de duración a dosis de 20, 40 y 80 mg una vez al día. Se estudió a un total de 5941 pacientes (3672 administrados con EDARBI®, 801 con placebo y 1468 con comparador activo) con hipertensión leve, moderada. En total, 51% de los pacientes fueron hombres y 26% tenían 65 años o más; 67% eran blancos y 19% negros. Dos estudios de 6 semanas soportan la eficacia de EDARBI® en la reducción de la presión sanguínea en comparación con placebo y comparadores activos. En el estudio 1, los pacientes fueron asignados aleatoriamente a placebo, EDARBI® 40 u 80 mg, olmesartán medoxomilo 40 mg o valsartán 320 mg una vez al día durante 6 semanas. Se midió la presión sanguínea clínica en posición sentada, en el punto más bajo, en la línea basal y cada dos semanas. En la semana 6, EDARBI® de 40 y 80 mg redujo la presión sanguínea clínica en posición sentada, en el punto más bajo, más que placebo como se muestra en la Figura 1.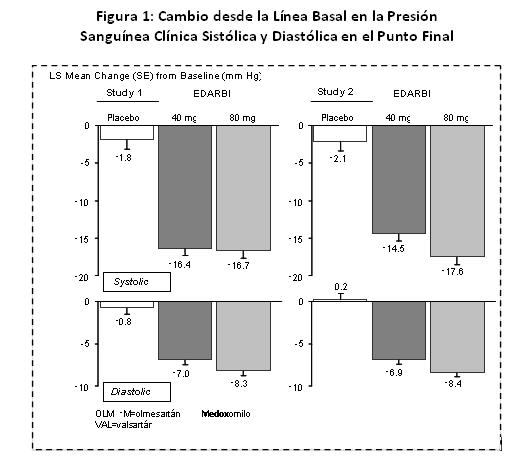
Como muestra la Figura 2, la mayor parte del efecto antihipertensivo ocurrió dentro de las primeras 2 semanas de dosificación con el efecto completo alcanzado a las 4 semanas y mantenido hasta el final del estudio.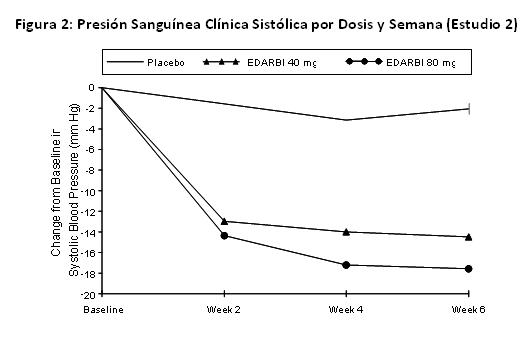
EDARBI® 40mg y 80 mg demostró reducciones estadísticamente significativas mayores que placebo en el criterio de valoración primario, cambio en la presión sanguínea sistólica media a las 24 horas mediante monitoreo ambulatorio de presión sanguínea a 6 semanas. Para el estudio 1, las reducciones en la presión sanguínea sistólica media a 24 horas fueron 0.3 mm Hg para placebo, 13.4 mm Hg para 40 mg y 14.5 mm Hg para 80 mg. Para el estudio 2, las reducciones en la presión sanguínea sistólica media a 24 horas fueron 1.4 mm Hg para placebo, 13.5 mm Hg para 40 mg y 14.6 mm Hg para 80 mg. El efecto de reducción de la presión sanguínea sistólica y diastólica de EDARBI® se mantuvo a lo largo de todo el intervalo de 24 horas de dosificación en todos los estudios controlados. Las mediciones ambulatorias por hora de la presión sanguínea sistólica del estudio 1 se muestran en la Figura 3. Las proporciones corregidas por placebo de cresta a valle para la presión sanguínea sistólica y diastólica fueron aproximadamente 80% o mayores en los Estudios 1 y 2.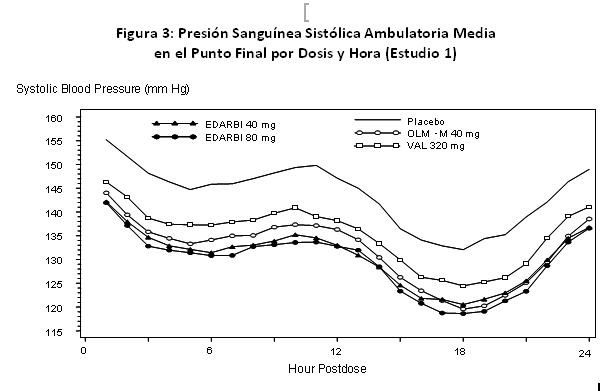
Los estudios 1 y 2 también compararon EDARBI® con la dosis aprobada más alta de olmesartán medoxomilo. En ambos estudios, EDARBI® 80 mg fue superior a olmesartán medoxomilo 40 mg con base en el criterio de valoración primario, cambio en la presión sanguínea sistólica media a 24 horas medida mediante monitoreo ambulatorio de la presión sanguínea. Se observaron reducciones de 2.5 y 2.1 mm Hg mayores que olmesartán medoxomilo en los Estudios 1 y 2, respectivamente. Se comparó EDARBI® con la dosis más alta aprobada de valsartán en dos estudios controlados. Éstos fueron el Estudio 1, como se describe antes y el Estudio 3 en el que los pacientes fueron asignados aleatoriamente para recibir EDARBI® 40 u 80 mg o valsartán 320 mg, respectivamente, durante un total de 6 meses. En ambos estudios, EDARBI® 80 mg fue superior a valsartán 320 mg, con base en el criterio de valoración primario, cambio en la presión sanguínea sistólica media a 24 horas medida mediante monitoreo ambulatorio de presión sanguínea. Se observaron reducciones de 4.3 y 4.0 mm Hg mayores que valsartán en los Estudios 1 y 3, respectivamente. El efecto de reducción de la presión sanguínea de EDARBI® contra placebo, olmesartán medoxomilo y valsartán con base en el criterio de valoración primario de presión sanguínea sistólica media a 24 horas se muestra en la Tabla 2.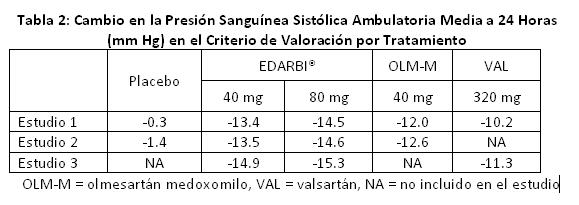
EDARBI® tiene un efecto antihipertensivo sostenido y consistente durante el tratamiento a largo plazo. No se observó efecto de rebote después de la interrupción abrupta de la terapia con EDARBI® después de 6 meses de tratamiento. EDARBI® fue efectivo en reducir la presión sanguínea independientemente de la edad, género o raza. El efecto fue algo menor en pacientes negros quienes tienden a tener menores niveles de renina. Esto es cierto en general para otros antagonistas de angiotensina II e inhibidores ACE. EDARBI® 40 y 80 mg administrado conjuntamente con un bloqueador del canal de calcio (amlodipino) o un diurético tipo tiazida (clortalidona) resultó en reducciones adicionales de la presión sanguínea. En un estudio clínico controlado, EDARBI® 40 y 80 mg coadministrado con amlodipino 5 mg resultó en reducciones de la presión sanguínea clínica sistólica y diastólica de 27.0/12.0 y 25.5/12.7 mm Hg, respectivamente, contra 15.9/7.1 mm Hg con amlodipino solo, En un estudio separado, EDARBI® 40 y 80 mg coadministrado con clortalidona 25 mg resultó en reducciones de la presión sanguínea clínica sistólica y diastólica de 36.2/16.2 y 34.4/16.0 mm Hg, respectivamente, contra 21.8/8.9 mm Hg con clortalidona sola.
Dosis y vía de administración: Dosis Recomendada: La dosis inicial recomendada es de 40 mg u 80 mg de azilsartán medoxomilo una vez al día con base a las cifras de presión arterial basal. La dosis máxima es de 80 mg de azilsartán medoxomilo una vez al día. EDARBI® puede tomarse con o sin alimentos. Poblaciones Especiales: No se recomienda ajustar la dosis inicial para pacientes ancianos, pacientes con insuficiencia renal de leve a moderada y en enfermedad renal en última etapa o con disfunción hepática de leve a moderada. EDARBI® no ha sido estudiado en pacientes con insuficiencia hepática severa. Uso Pediátrico: No se ha establecido la seguridad y eficacia en pacientes pediátricos menores de 18 años de edad. Uso Geriátrico: No es necesario el ajuste de dosis con EDARBI® en pacientes ancianos. Del total de pacientes en estudios clínicos con EDARBI®, 26% fueron ancianos (65 años de edad o mayores), 5% tenían 75 años de edad o más. No se observaron diferencias generales en la seguridad o eficacia entre los pacientes ancianos y los pacientes jóvenes, pero no se puede descartar una mayor sensibilidad de algunos individuos mayores. Insuficiencia Renal: No se requiere ajuste de dosis en pacientes con insuficiencia renal de leve a severa o enfermedad renal de última etapa. La hemodiálisis no elimina a azilsartán de la circulación sistémica. Insuficiencia Hepática: No se requiere ajuste de dosis en sujetos con insuficiencia hepática de leve o moderada. EDARBI® no ha sido estudiado en pacientes con insuficiencia hepática severa.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: Hay pocos datos relacionados con la sobredosis en humanos. Durante los estudios clínicos controlados en sujetos saludables, se administraron dosis de hasta 320 mg una vez al día durante 7 días y fueron bien tolerados. En el caso de una sobredosis, se deberá instituir terapia de apoyo según lo dicte el estatus clínico del paciente. Azilsartán no es dializable.
Presentación(es): Caja de cartón con blíster de 7, 10, 14, 20, 28 y 30 tabletas de 40 y 80 mg Frasco con 7, 14 y 28 tabletas de 40 y 80 mg
Leyendas de protección: Literatura exclusiva para médicos. Su venta requiere receta médica. Prohibida la venta fraccionada del producto. No se deje al alcance de los niños. No se use durante el embarazo, lactancia y menores de 18 años. No se administre simultáneamente con aliskireno ni con inhibidores de la enzima convertidora de angiotensina. Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx
Nombre y domicilio del laboratorio: Takeda México S.A de C.V. Av. Primero de Mayo No. 130. Col. Industrial Atoto, CP 53519. Naucalpan de Juárez, México, México.
Número de registro del medicamento: 106M2011 SSA IV