ELOMAZIV®
SYNTHON
Denominación genérica: Bortezomib.
Forma farmacéutica y formulación: Solución. Cada frasco vial con polvo liofilizado contiene: Ester borónico de manitol equivalente a 3.5 mg de Bortezomib. Excipiente cs.
Indicaciones terapéuticas: Reconstituir con 3.5 ml de solución inyectable de Cloruro Sódico al 0.9% ELOMAZIV® está indicado como parte del tratamiento de combinación de pacientes con mieloma múltiple no tratados previamente. ELOMAZIV® está indicado en el tratamiento del mieloma múltiple en pacientes que han recibido al menos una terapia previa. ELOMAZIV® está indicado en el tratamiento de pacientes con linfoma de células del manto que han recibido al menos una terapia previa.
Farmacocinética y farmacodinamia: Mecanismo de acción: El ingrediente activo de ELOMAZIV®: bortezomib, es un inhibidor reversible de la actividad similar a la quimiotripsina del proteosoma 26S en células de mamífero. EI proteosoma 26S es un gran complejo proteico que degrada a las proteínas ubiquitinizadas. La ruta proteosoma-ubiquitina juega un papel esencial en la regulación de la concentración intracelular de proteínas específicas, manteniendo así la homeostasis de la célula. La inhibición del proteosoma 26S evita la proteólisis dirigida, la cual puede afectar la activación de múltiples cascadas de señalización dentro de la célula. Esta interrupción del mecanismo normal de la homeostasis puede llevar a la muerte celular. Experimentos in vitro han demostrado que el ingrediente activo de ELOMAZIV® es citotóxico para una gran variedad de células cancerosas y retrasa el crecimiento del tumor in vivo en modelos no clínicos de tumores, incluyendo el mieloma múltiple. Estudio clínico aleatorizado, abierto en pacientes con mieloma múltiple previamente no tratados: Se realizó un estudio prospectivo, fase 3, internacional, aleatorizado (1:1), abierto de 682 pacientes para determinar si ELOMAZIV® (1.3 mg/m2) en combinación con melfalán (9 mg/m2) y prednisona (60 mg/m2) dio como resultado una mejoría en el tiempo a la progresión (TTP) cuando se comparó con melfalán (9 mg/m2) y prednisona (60 mg/m2) en pacientes con mieloma múltiple previamente no tratados. El tratamiento se administró por un máximo de 9 ciclos (aproximadamente 54 semanas) y se suspendió tempranamente por progresión de la enfermedad o toxicidad inaceptable.
Farmacocinética: La mediana estimada de la concentración máxima plasmática obtenida después de la administración intravenosa de una dosis de ELOMAZIV® de 1.3 mg/m², fue de 509 ng/ml (rango de 109 a 1,300 ng/ml) en ocho pacientes con mieloma múltiple y los valores de depuración de creatinina se encontraron en el rango de 31-169 ml/min. En pacientes con enfermedad tumoral maligna avanzada, la vida media de eliminación fue de 9 a 15 horas después de la primera administración, con un esquema de dosis en rango de 1.45 a 2.00 mg/m². La farmacocinética de la dosis recomendada de ELOMAZIV® como agente único en pacientes con mieloma múltiple no se ha caracterizado. Distribución: El volumen de distribución de ELOMAZIV® como agente único no ha sido evaluada en las dosis recomendadas en pacientes con mieloma múltiple. La unión a proteínas plasmáticas de bortezomib fue de 83% en promedio, con un rango de concentración de 100-1,000 ng/ml. Metabolismo: Estudios in vitro con microsomas de hígado humano y con fragmentos de cADN que codifican para isoenzimas del citocromo P-450 indican que bortezomib se metaboliza principalmente en forma oxidativa por las enzimas del citrocromo P-450, 3A4, 2C19 y 1A2, el metabolismo de bortezomib por CYP 2D6 y 2C9 es menor. La principal ruta metabólica es la deboronación que da lugar a dos metabolitos deboronados, que posteriormente sufren hidroxilación. Los metabolitos deboronados de bortezomib son inactivos como inhibidores del proteosoma 26S. Los datos obtenidos de muestras de plasma de 8 pacientes, tomadas a los 10 y 30 minutos después de la dosis, indican que los niveles plasmáticos de los metabolitos son bajos comparados con los niveles del fármaco principal. Eliminación: Las vías de eliminación de ELOMAZIV® no han sido caracterizadas en seres humanos. Poblaciones especiales: Edad, género y raza: No se han evaluado los efectos de la edad, género y raza sobre la farmacocinética de ELOMAZIV®. Daño hepático: No se condujeron estudios de farmacocinética en pacientes con daño hepático. Daño renal: No se condujeron estudios de farmacocinética en pacientes con daño renal. Los estudios clínicos incluyeron pacientes con depuración de creatinina con valores en el rango de 13.8-220 ml/min. Pediátricos: No existen datos disponibles de la farmacocinética de ELOMAZIV® en niños. Geriátricos: No se observaron diferencias en la seguridad o efectividad entre los pacientes > 65 años y los jóvenes que han recibido ELOMAZIV® pero no se puede descartar mayor sensibilidad en algunos individuos mayores.
Contraindicaciones: Hipersensibilidad al ingrediente activo, al boro o al manitol.
Precauciones generales: ELOMAZIV® debe administrarse bajo la supervisión de personal experimentado en el uso de terapia antineoplásica. En general, el perfil de seguridad de los pacientes tratados con ELOMAZIV® como monoterapia fue similar al observado en los pacientes tratados con ELOMAZIV® en combinación con melfalán y prednisona. Neuropatía periférica: El tratamiento con ELOMAZIV® causa neuropatía periférica, predominantemente sensitiva, aunque también se han reportado casos de neuropatía mixta: sensitiva-motora. Los pacientes con síntomas preexistentes (entumecimiento, dolor o sensación quemante en pies y manos) y/o signos de neuropatía periférica pueden experimentar empeoramiento de la neuropatía periférica (incluyendo ≥ grado 3) durante el tratamiento con ELOMAZIV® y deben monitorizarse vigilando los síntomas de neuropatía, tales como; sensación quemante, hiperestesia, hipoestesia, parestesia, malestar o dolor neuropático. Los pacientes que experimenten nueva neuropatía periférica o en los que empeore, pueden requerir cambio en la dosis y esquema de tratamiento de ELOMAZIV® Después del ajuste de dosis se reportó una mejoría o la resolución de la neuropatía periférica en el 51% de los pacientes con neuropatía periférica grado ≥ 2 en el estudio de mieloma múltiple fase 3 con monoterapia y en 73% de los pacientes que interrumpieron el tratamiento debido a neuropatía grado 2 o neuropatía periférica grado ≥ 3 en los estudios de mieloma múltiple fase 2. No se han realizado estudios a largo plazo de neuropatía periférica en linfomas de células del manto. Hipotensión: En estudios de mieloma múltiple fase 2 y 3 con monoterapia la incidencia de hipotensión (postural, ortostática e hipotensión NOS) fue de 11% a 12%. Estos eventos se observan durante la terapia. Debe tenerse precaución cuando se trate a pacientes con antecedentes de síncope, pacientes que reciben medicamentos asociados con hipotensión y pacientes deshidratados. El manejo de la hipotensión ortostática/postural puede incluir el ajuste de medicamentos antihipertensivos hidratación y administración de mineralocorticoides y/o simpaticomiméticos. Alteraciones cardiacas: Se ha observado el desarrollo agudo o la exacerbación de falla cardiaca congestiva, y/o disminución de la fracción de eyección ventricular izquierda de nuevo inicio, incluyendo reportes de pacientes con pocos o ningún factor de riesgo para disminución de la fracción de eyección ventricular izquierda. Los pacientes con enfermedades cardiacas o con factores de riesgo deben vigilarse estrechamente. En el estudio de mieloma múltiple fase 3 con monoterapia, la incidencia para cualquier desorden cardiaco emergente del tratamiento fue de 15% y de 13%, en los grupos con ELOMAZIV® y dexametasona, respectivamente. La incidencia de eventos de falla cardiaca (edema pulmonar agudo, falla cardiaca, falla cardiaca congestiva, choque cardiogénico y edema pulmonar) fue similar en los grupos con ELOMAZIV® y dexametasona, 5% y 4%, respectivamente. En los estudios clínicos ha habido casos aislados de prolongación del intervalo QT, no se ha podido establecer la causalidad. Eventos hepáticos: Raramente se han reportado casos de falla hepática aguda en los pacientes que están recibiendo múltiples medicamentos concomitantemente y con una condición médica delicada. Otros eventos hepáticos reportados incluyen elevación de las enzimas hepáticas, hiperbilirrubinemia y hepatitis. Tales cambios son reversibles una vez que se suspende ELOMAZIV®. Existe limitada información con respecto a estos pacientes. Alteraciones pulmonares: Raramente se han reportado casos de enfermedad pulmonar infiltrativa difusa aguda de etiología desconocida, tales como: pneumonitis, neumonía intersticial, infiltración pulmonar y Síndrome de Distres Respiratorio Agudo (SDRA) en pacientes que se encuentran recibiendo ELOMAZIV®. Algunos de estos eventos han sido fatales. Una alta proporción de estos eventos ha sido reportada en Japón. En caso de nuevos eventos o empeoramiento de síntomas pulmonares, se debe realizar una pronta evaluación diagnóstica y dar tratamiento apropiado a los pacientes. Exámenes de laboratorio: Se debe monitorear frecuentemente, durante el tratamiento con ELOMAZIV® la cuenta de sangre completa. Trombocitopenia: ELOMAZIV® se asocia a trombocitopenia. Las plaquetas se encontraban más bajas en el día 11 de cada ciclo de tratamiento con ELOMAZIV® recuperándose el valor inicial en el siguiente ciclo. El patrón cíclico de disminución y recuperación de la cuenta plaquetaria permaneció constante durante los 8 ciclos de dos dosis semanalmente y no hubo evidencia de trombocitopenia acumulativa. El promedio de la cuenta plaquetaria más baja medida fue aproximadamente el 40% de la cuenta basal. La severidad de la trombocitopenia relacionada a la cuenta plaquetaria pretratamiento se muestra en la tabla 1 para el estudio fase 3 con monoterapia. La incidencia de eventos de sangrados significativos (≥ grado 3), en el estudio de mieloma múltiple fase 3 con monoterapia fue similar tanto en el brazo con ELOMAZIV® (4%) como en el de dexametasona (5%). La cuenta plaquetaria debe monitorearse antes de cada dosis de ELOMAZIV®. El tratamiento con ELOMAZIV® debe suspenderse cuando la cuenta plaquetaria sea < 25,000/ml y reiniciarse con una dosis reducida. Ha habido reportes de hemorragia gastrointestinal e intracerebral asociada con ELOMAZIV®. Las transfusiones pueden realizarse a criterio del médico.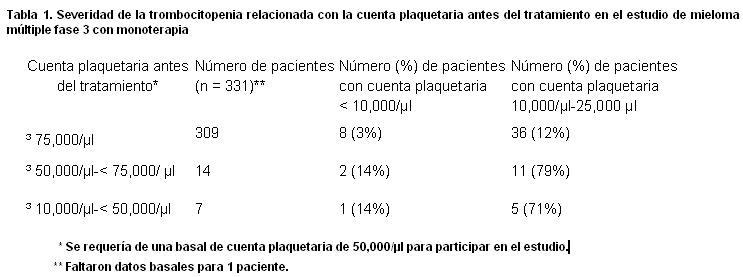
Eventos adversos gastrointestinales: El tratamiento con ELOMAZIV®, puede causar náuseas, diarrea, constipación y vómito, requiriéndose en ocasiones el uso de antieméticos o antidiarreicos. Para tratar la deshidratación se deben administrar líquidos y electrólitos. Debido a que los pacientes que están recibiendo tratamiento con ELOMAZIV®, pueden tener vómito y/o diarrea, se les debe orientar con respecto a medidas apropiadas para evitar deshidratación. Se les debe comentar que en caso de presentar mareo, cefalea o desvanecimiento deben ponerse en contacto con su médico. Síndrome de lisis tumoral: Pueden ocurrir complicaciones de síndrome de lisis tumoral debido a que ELOMAZIV® .es un agente citotóxico que mata rápidamente células malignas. Los pacientes en riesgo de síndrome de lisis tumoral, son aquéllos con un alto volumen de masa tumoral antes del tratamiento. Estos pacientes deben vigilarse de cerca y deben tomarse precauciones apropiadas. Pacientes con daño hepático: Bortezomib es metabolizado por las enzimas hepáticas, por lo que su eliminación puede disminuir en pacientes con daño hepático. Estos pacientes deben monitorizarse estrechamente para toxicidades cuando son tratados con ELOMAZIV® .
Efectos sobre la habilidad para manejar y utilizar maquinaria: Los pacientes deben ser cuidadosos al operar maquinaria o manejar automóviles, ya que ELOMAZIV® .puede causar disminución de la presión sanguínea así como también, cansancio, mareo, desvanecimiento o visión borrosa.
Restricciones de uso durante el embarazo y la lactancia: No se use durante el embarazo y la lactancia. En caso de mujeres en edad reproductiva debe evitarse el embarazo durante el tratamiento con ELOMAZIV®. Se desconoce si el ingrediente activo se excreta en la leche materna, y al igual que ocurre con el uso de muchos medicamentos, debe evitarse la lactancia durante el tratamiento con ELOMAZIV® debido al potencial de reacciones adversas graves reportadas en los lactantes. En estudios no clínicos de toxicidad en ratas y conejos, ELOMAZIV® no fue teratogénico con la dosis mayor probada [0.075 mg/kg (0.5 mg/m²) en la rata y 0.05 mg/kg (0.6 mg/m²)] en el conejo durante la organogénesis. Estas dosis son aproximadamente la mitad de la dosis clínica de 1.3 mg/m² de acuerdo al área de la superficie corporal. Se administró ELOMAZIV® a conejas encintas durante la organogénesis a una dosis de 0.05 mg/kg (0.6 mg/m²) donde se observó una significativa pérdida y disminución del número de fetos vivos postimplantación. Los fetos vivos de estas camadas también mostraron una disminución considerable en el peso fetal. La dosis es aproximadamente 0.5 veces la dosis clínica que es de 1.3 mg/m², la cual se basa en el área de la superficie corporal. No se realizaron estudios con ELOMAZIV® de transferencia de placenta. No hay estudios adecuados y con un buen control en mujeres embarazadas. Si ELOMAZIV® se usa durante el embarazo o si la paciente se embaraza durante el tratamiento con el fármaco, se debe informar a la paciente del riesgo potencial para el feto. Las pacientes deben ser advertidas del uso de medidas contraceptivas efectivas para prevenir el embarazo y evitar la lactancia durante el tratamiento con ELOMAZIV®. Lactancia: Se desconoce si ELOMAZIV® es excretado o no por la leche materna humana. Debido a que muchos medicamentos son excretados en la leche y por la potencial reacción adversa seria que existe con ELOMAZIV® al amamantar a los infantes, las mujeres deben ser advertidas que no deben amamantar mientras están bajo tratamiento con ELOMAZIV®.
Reacciones secundarias y adversas: Resumen de los estudios clínicos en pacientes con mieloma múltiple: La seguridad y eficacia de ELOMAZIV® fue evaluada en 3 estudios con la dosis recomendada de 1.3 mg/m². Éstos incluyen un estudio fase 3, aleatorizado, comparativo vs. dexametasona de 669 pacientes con mieloma múltiple con recaída o refractario que hubieran recibido 1-3 líneas de tratamiento anterior (M34101-039); un estudio fase 2 multicéntrico, abierto, de un brazo de 202 pacientes que hubieran recibido por lo menos 2 líneas de tratamiento anterior y que tuvieran progresión de la enfermedad con el tratamiento más reciente (M34100-025) y un estudio fase 2 de respuesta clínica a las dosis de 1.0 mg/m² o 1.3 mg/m² de ELOMAZIV® en pacientes con mieloma múltiple en recaída que hayan tenido progresión o recaída después de un tratamiento de primera línea (M34100-024).
Resumen de los estudios clínicos realizados en pacientes con mieloma múltiple sin tratamiento previo: La siguiente tabla describe los datos de seguridad de 340 pacientes con mieloma múltiple Naives a tratamiento que recibieron ELOMAZIV® (1.3 mg/m2) en combinación con melfalán (9 mg/m2) y prednisona (60 mg/m2), estudio fase 3, prospectivo.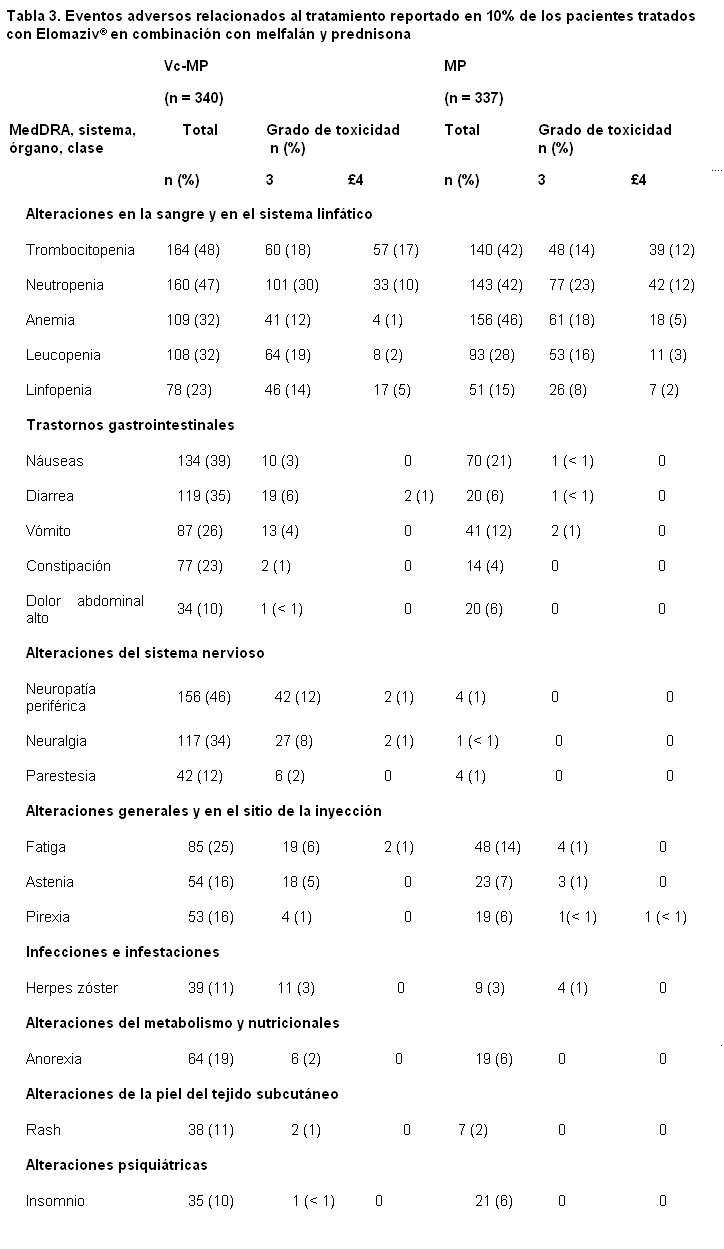
Reactivación del virus del herpes zóster: En el estudio fase 3 en pacientes con mieloma múltiple no tratados previamente, 27% de los pacientes recibieron antivirales profilácticamente, en el brazo de VcMP. En este estudio la reactivación del herpes zoster fue más común en los pacientes con VcMP comparado con MP (11% vs 3% respectivamente). Tres (3%) de los 91 pacientes en el grupo de tratamiento con VcMP quienes recibieron antivirales como medida profiláctica, tuvieron una reactivación de la infección por herpes zóster. Pacientes con linfoma de células del manto: Se evaluaron los datos de seguridad de pacientes con linfoma de células del manto en un estudio fase 2, en el que se incluyeron 155 pacientes tratados con ELOMAZIV® a la dosis recomendada de 1.3 mg/m2. El perfil de seguridad de ELOMAZIV® en estos pacientes fue similar a la observada en los pacientes con mieloma múltiple. Las diferencias notables entre estas dos poblaciones de pacientes fueron la trombocitopenia, neutropenia, anemia, náusea, vómito y pirexia, las cuales fueron reportadas con mayor frecuencia en los pacientes con mieloma múltiple que en los pacientes con linfoma de células del manto; la neuropatía periférica, el rash y prurito fueron mayores en los pacientes con linfoma de células del manto en comparación con los pacientes con mieloma múltiple. Experiencia post-comercialización: Aquí se enlistan las reacciones adversas al medicamento clínicamente significativas si no se han mencionado anteriormente. Las frecuencias proporcionadas abajo reflejan las tasas de reacciones adversas observadas mundialmente después de la comercialización de ELOMAZIV®. Las reacciones adversas del medicamento están clasificadas por frecuencia, empleando los siguientes principios: Muy común ( > 1/10), común ( > 1/100 y < 1/10), poco común ( > 1/1,000 y < 1/100), raro ( > 1/10,000 y < 1/1,000), muy raro ( < 1/10,000, incluyendo reportes aislados).
Investigaciones ECG: Ha habido casos aislados de prolongación del intervalo QT en estudios clínicos, la causalidad no se ha establecido bien. Reacciones en la piel: Se presentó rash en el 21% de los pacientes.
Interacciones medicamentosas y de otro género: Estudios in vitro y ex vivo en animales indican que el ingrediente activo de ELOMAZIV® es un inhibidor débil de las isoenzimas del citrocromo (CYP) P-450 1A2, 2C9, 2C19, 2D6 y 3A4. Debido a la limitada contribución (7%) de CYP2D6 en el metabolismo de ELOMAZIV® no se espera que el fenotipo metabolizador pobre afecte la disposición total de ELOMAZIV®. ELOMAZIV® no induce las actividades del citrocromo P-450 3A4 y 1A2 en cultivo primario de hepatocitos humanos. En un estudio de interacción medicamentosa se evaluó el efecto del ketoconazol, un potente inhibidor de la CYP3A, el cual mostró un incremento promedio del 35% del ABC de ELOMAZIV® con base en la información de 12 pacientes. Por lo que los pacientes que estén recibiendo ELOMAZIV® en combinación con algún inhibidor potente de la CYP3A4 (p. ej., ketokonazol ritonavir, etc.) deben de ser monitoreados muy de cerca. En un estudio de interacción medicamentosa en el que se evaluó el efecto de omeprazol, inhibidor potente de la CYP2C19, no se observó efecto significativo alguno en la farmacocinética de ELOMAZIV® en 17 pacientes. En otro estudio de interacción medicamentosa se observó que el efecto de melfalán-prednisona sobre ELOMAZIV® fue un incremento promedio del 17% del ABC de ELOMAZIV®, 21 pacientes. Esto no se considera clínicamente relevante. Durante los estudios clínicos, se reportaron casos de hipoglucemia e hiperglucemia en pacientes diabéticos que recibían hipoglucemiantes orales. Los pacientes tratados con medicamentos orales antidiabéticos, que reciben tratamiento con ELOMAZIV® requieren vigilancia estrecha de los niveles de glucosa en sangre y pueden requerir el ajuste de la dosis de su tratamiento antidiabético. Debido a los eventos de hipotensión reportados durante los estudios clínicos, puede ser necesario ajustar la dosis de medicamentos antihipertensivos en pacientes tratados con ELOMAZIV®. Los pacientes deben ser advertidos del uso concomitante de medicamentos que pueden asociarse con neuropatía periférica (tales como amiodarona, antivirales, isoniacida, nitrofurantoína o estatinas) o con disminución de la presión arterial.
Alteraciones en los resultados de pruebas de laboratorio: Durante el tratamiento con ELOMAZIV® deben evaluarse los parámetros de la biometría hemática completa, incluyendo el conteo de plaquetas.
Precauciones en relacion con los efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: ELOMAZIV® mostró actividad clastogénica (aberraciones cromosómicas estructurales) en los ensayos in vitro de aberración cromosómica con células ováricas de hámster. ELOMAZIV® No mostró genotoxicidad en las pruebas de mutagenicidad in vitro e in vivo en ensayos de micronúcleos en ratones. ELOMAZIV® podría tener un potencial efecto en la fertilidad masculina y femenina. La evaluación de tejidos reproductivos se llevó a cabo en los estudios de toxicidad general pues aún no se realizan estudios específicos sobre la fertilidad. En un estudio de toxicidad en ratas, con duración de 6 meses, se observaron efectos degenerativos en el ovario a dosis ≥ 0.3 mg/m² (un cuarto de la dosis clínica inicial recomendada) y cambios degenerativos en las pruebas ocurrieron a dosis de 1.2 mg/m². No se han llevado a cabo estudios de carcinogénesis con ELOMAZIV®.
Dosis y via de administración: I.V. Monoterapia: La dosis recomendada de ELOMAZIV® es de 1.3 mg/m²/dosis administrado como bolo intravenoso de 3 a 5 segundos dos veces por semana durante dos semanas (días 1, 4, 8 y 11) seguido por un periodo de descanso de 10 días (días 12 a 21). Estas 3 semanas se consideran un ciclo de tratamiento, para extender la terapia a más de 8 ciclos, ELOMAZIV® puede administrarse en un esquema estándar o en un programa de mantenimiento de una vez por semana por 4 semanas (días 1, 8, 15 y 22) seguido por un periodo de descanso de 13 días (días 23 a 35). Deben de transcurrir por lo menos 72 horas entre dosis consecutivas. Modificación de dosis o reinicio del tratamiento: Debe suspenderse la terapia con ELOMAZIV® al inicio de cualquier toxicidad grado 3 no-hematológica o toxicidad hematológica grado 4 excluyendo la neuropatía. Una vez que los síntomas de la toxicidad se hayan resuelto puede iniciarse la terapia a una dosis reducida del 25% (1.3 mg/m²/dosis reducida a 1.0 mg/m²/dosis; 1.0 mg/m²/dosis a 0.7 mg/m²/dosis). La modificación de dosis recomendada para el manejo de pacientes que experimenten dolor neuropático y/o neuropatía sensitiva periférica relacionada con el tratamiento con ELOMAZIV® está contenida en la siguiente tabla:
Criterios Comunes de Toxicidad del Instituto Nacional de Cáncer de Estados Unidos. Antes de iniciar el tratamiento con ELOMAZIV® en pacientes con neuropatía severa preexistente es necesario valorar cuidadosamente el riesgo beneficio del tratamiento. Pacientes con insuficiencia renal: La farmacocinética de ELOMAZIV® no es modificada por el grado de insuficiencia renal. Por lo que no es necesario ajustar la dosis de ELOMAZIV® en los pacientes con insuficiencia renal. Ya que la diálisis puede reducir las concentraciones de ELOMAZIV® el medicamento debe administrarse después de haberse realizado la diálisis. Se llevó a cabo un estudio en pacientes con varios grados de insuficiencia renal, se clasificaron de acuerdo a los valores de la depuración de creatinina (CrCL) en los siguientes grupos: normal (CrCL 60 ml/min/1.73 m2, n = 12), leve (CrCL = 40-59 ml/min/1.73 m2, n = 10), moderada (CrCL = 20-39 ml/min/1.73 m2, n = 9), y severa (CrCL < 20 ml/min/1.73 m2, n = 3). Se incluyó un grupo de pacientes que estaban con diálisis, se les administró el medicamento después de ser dializados (n = 8). A los pacientes se les administró una dosis intravenosa de 0.7 a 1.3 mg/m2 de ELOMAZIV® dos veces por semana. La exposición a ELOMAZIV® fue comparable entre todos los grupos (normalización de la dosis por ABC y Cmáx). Vía de administración: Intravenosa. ELOMAZIV® Se administra como bolo intravenoso de 3-5 segundos, a través de un catéter central o periférico, seguido de un flujo de solución isotónica inyectable de cloruro de sodio. Antes de usarse el polvo liofilizado de ELOMAZIV® se reconstituye con 3.5 ml de solución inyectable de cloruro de sodio (0.9%). La solución resultante debe ser clara e incolora. Una vez reconstituido y antes de aplicarse debe inspeccionarse en búsqueda de partículas o decoloraciones, en caso de ser positiva la búsqueda no debe emplearse el producto reconstituido. Terapia de combinación: Dosis recomendada: ELOMAZIV® es administrado como bolo intravenoso de 3-5 segundos en combinación con melfalán oral y prednisona oral por nueve ciclos de tratamiento de 6 semanas como se muestra en la tabla 6. En los ciclos 1-4, ELOMAZIV® es administrado dos veces a la semana (días 1, 4, 8, 11, 22, 25, 29 y 32). En los ciclos 5-9, ELOMAZIV® se administra una vez a la semana (días 1, 8, 22 y 29).
Guías de manejo de dosis en el tratamiento de combinación: Modificación de la dosis y reinicio de tratamiento cuando ELOMAZIV® es administrado en combinación con melfalán y prednisona: Antes de iniciar un nuevo ciclo de tratamiento: La cuenta plaquetaria debe ser 70 x 109/L y la ANC debe ser 1.0 x 109/L. Las toxicidades no hematológicas debieron resolverse, regresar a la línea basal o a grado 1.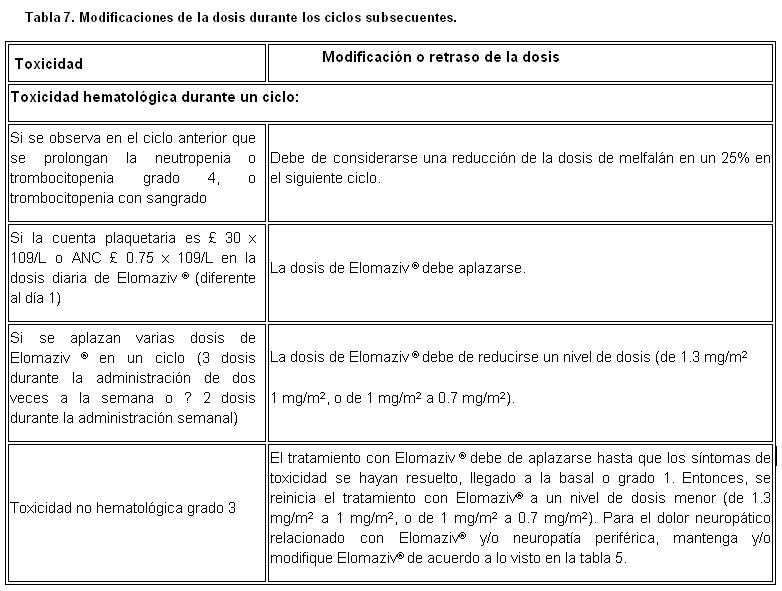
Para información adicional con respecto a melfalán y a prednisona, favor de ver la información para prescribir de estos productos. Precauciones para la administración: ELOMAZIV® es un antineoplásico y debe manejarse con precaución durante su reconstitución y administración. Se recomienda utilizar una técnica aséptica apropiada, usar guantes y ropa protectora para prevenir el contacto con la piel. En los estudios clínicos, se reportó un 5% de irritación de la piel local, pero la extravasación de ELOMAZIV® no se asoció con daño del tejido. Cualquier material no empleado o de desecho debe desecharse de acuerdo a los requerimientos locales
Manifestaciones y manejo de la sobredosificación o ingesta accidental: Los estudios farmacológicos de seguridad cardiovascular en monos y perros demuestran que la dosis intravenosa (IV), aproximadamente 2 a 3 veces la dosis clínica inicial recomendada en mg/m2, se asocia con un incremento en la frecuencia cardiaca, disminución en la contractibilidad, hipotensión y muerte. La disminución en la contractibilidad e hipotensión responde a la intervención aguda con un inotrópico positivo o agente vasopresor. En estudios en perros se observó un ligero incremento del intervalo QT a dosis letales. En pacientes, sobredosificar más de dos veces la dosis recomendada se ha asociado con el inicio agudo de hipotensión sintomática y trombocitopenia con resultados fatales. Se desconoce un antídoto específico para la sobredosificación con ELOMAZIV®. En un evento de sobredosificación, deben monitorearse los signos vitales del paciente y administrar terapia de soporte para mantener la presión sanguínea (tales como fluidos, vasopresores y/o agentes inotrópicos) y la temperatura corporal.
Presentación(es): Caja con un frasco vial con 3.5 mg de polvo liofilizado para reconstituir e instructivo anexo.
Recomendaciones sobre almacenamiento: Consérvese a no más de 25 °C. Protéjase de la luz. Una vez reconstituido el medicamento debe conservarse a no más de 25°C y protegido de la luz. Consérvese en su empaque original.
Leyendas de protección: Literatura exclusiva para médicos. No se administre si la solución reconstituida no es transparente, si contiene partículas en suspensión o sedimentos. Una vez reconstituido este medicamento debe ser administrado inmediatamente. Si no se usa inmediatamente en los tiempos de conservación y en las condiciones recomendadas, la responsabilidad es del usuario; sin embargo la solución reconstituida es estable dentro de las primeras 8 horas de preparación, durante este periodo de tiempo debe ser conservada en el frasco vial o en una jeringa de polipropileno estéril a una temperatura no mayor de 25°C y protegido de la luz. No se deje al alcance de los niños. No se use en el embarazo y la lactancia. Si no se administra todo el producto deséchese el sobrante. Reporte las sospechas de reacciones adversas a los correos: farmacovigilancia@cofepris.gob.mx y farmacovigilancia@synthon.com
Nombre y domicilio del laboratorio: Hecho en República Checa por:
Oncomed Manufacturing A.S. Karásek 2229/1b, budova 02, 621 00 Brno-Reckovice. República Checa. Acondicionado secundario por: GE Pharmaceuticals Ltd. Industrial Zone, Chekanitza-South Area, 2140, Botevgrad, Bulgaria. Distribuido por: SYNTHON MÉXICO, S. A. DE C. V. Periférico Sur No. 8565-A Bodega No. 2, Col. El Mante, C.P. 45609, Tlaquepaque, Jalisco, México. Acondicionado secundario por: SYNTHON MÉXICO, S. A. DE C. V. Justo Sierra No. 933. Col. Agua Blanca Industrial. C.P. 45235. Zapopan, Jalisco, México.
Número de registro del medicamento: 081M2016 SSA IV