EMTRIVA®
STENDHAL
Denominación genérica: Emtricitabina.
Forma farmacéutica y formulación: Cápsula. Cada cápsula de gelatina dura contiene: emtricitabina 200 mg. Excipiente cbp 1 cápsula.
Indicaciones terapéuticas: EMTRIVA® está indicado, en combinación con otros agentes antirretrovirales, para el tratamiento de la infección por VIH-1 en adultos. Información adicional importante sobre el uso de EMTRIVA® para el tratamiento de la infección por VIH-1: EMTRIVA® no deberá coadministrarse con ATRIPLA™ (una asociación de dosis fija de efavirenz, emtricitabina y tenofovir disoproxil fumarato), TRUVADA® o productos que contengan lamivudina (véase Precauciones generales). En pacientes que han recibido tratamiento antirretroviral previo, el uso de EMTRIVA® deberá estar guiado por análisis de laboratorio y los antecedentes de tratamiento (véase Farmacocinética y farmacodinamia). Estudio clínicos: pacientes adultos sin tratamiento previo: estudio 934: se han reportado los resultados a 144 semanas del estudio 934, un ensayo clínico comparativo, abierto, aleatorizado, en el cual se compara la combinación de VIREAD® + efavirenz contra la tableta única de zidovudina/lamivudina + efavirenz en 511 pacientes naïve. Desde la 96ª a la 144ª semana del estudio, los pacientes recibieron emtricitabina + tenofovir disoproxil fumarato (tenofovir DF) con efavirenz, en lugar de EMTRIVA® + tenofovir DF con efavirenz. Los pacientes tuvieron una edad media de 38 años (intervalo 18 a 80), 86% fueron varones, 59% caucásicos y 23% de raza negra. La cuenta basal mediana de células CD4+ fue de 245 cels/mm3 (intervalo 2-1191) y la media de la carga del RNA viral del VIH-1 en plasma (Carga Viral) fue de 5,01 log10 copias/ml (intervalo 3,56 a 6,54). Se estratificó a los pacientes de acuerdo con la cuenta inicial de linfocitos CD4+ ( < o ≥ 200 cels/mm3); 41% de los sujetos tuvo una cuenta de CD4+ ( < 200 cels/mm3); 51% de los pacientes tuvo una carga viral basal > 100.000 copias/ml. En la tabla 1, se presentan los resultados del tratamiento después de 48 y de 144 semanas de tratamiento en los pacientes que no presentaban resistencia al efavirenz al inicio.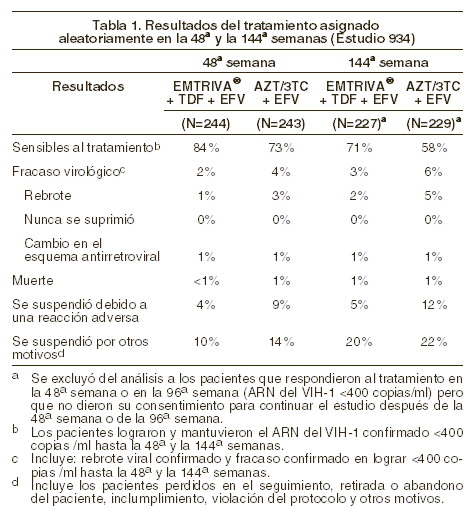
Hasta la 48ª semana, el 84% de los pacientes del grupo tratado con EMTRIVA® + tenofovir DF y el 73% de los pacientes tratados con zidovudina y lamivudina lograron y mantuvieron el ARN del VIH-1 < 400 copias/ml (hasta la 144ª semana: 71% y 58%, respectivamente). La diferencia en la proporción de pacientes que lograron y mantuvieron el ARN del VIH-1 < 400 copias/ml después de 48 semanas de tratamiento es principalmente el resultado del mayor número de interrupciones del tratamiento debidas a reacciones adversas y a otros motivos en el grupo tratado con zidovudina y lamivudina, en este estudio abierto. Además, el 80% de los pacientes del grupo tratado con EMTRIVA® + tenofovir DF y el 70% de los pacientes tratados con zidovudina y lamivudina lograron y mantuvieron el ARN del VIH-1 < 50 copias/ml hasta la 48ª semana (hasta la 144ª semana: 64% y 56%, respectivamente). En la 48ª semana, el aumento medio con respecto a los valores iniciales del recuento de linfocitos CD4+ fue de 190 linfocitos/mm3 en el grupo tratado con EMTRIVA® + tenofovir DF, y de 158 linfocitos/mm3 en el grupo que recibió zidovudina y lamivudina (en la 144ª semana: 312 y 271 linfocitos/mm3, respectivamente). A las 48 semanas, siete pacientes del grupo tratado con EMTRIVA® + tenofovir DF y cinco pacientes del grupo tratado con zidovudina y lamivudina experimentaron una nueva reacción de clase C, según el código de los CDC (10 y 6 pacientes, respectivamente, hasta las 144 semanas). Estudio 301 A: el estudio 301 A fue un estudio multicéntrico, controlado con substancia activa, doble ciego y con duración de 48 semanas, que comparó EMTRIVA® (200 mg una vez al día) administrado en combinación con didanosina y efavirenz versus estavudina, didanosina y efavirenz, en 571 pacientes naïve. La edad promedio de los pacientes fue de 36 años (intervalo de 18 a 69); 85% fueron hombres; 52% caucásicos, 16% afroamericanos y 26% hispanos. Los pacientes tuvieron un recuento celular CD4+ basal medio de 318 células/mm3 (intervalo 5-1317) y carga viral basal media de 4,9 log10 copias/ml (intervalo 2,6-7,0). Treinta y ocho por ciento de los pacientes tuvieron carga viral basal de > 100.000 copias/ml y 31% tuvieron un recuento celular CD4+ < 200 células/ml. Los resultados del tratamiento se muestran en la tabla 2.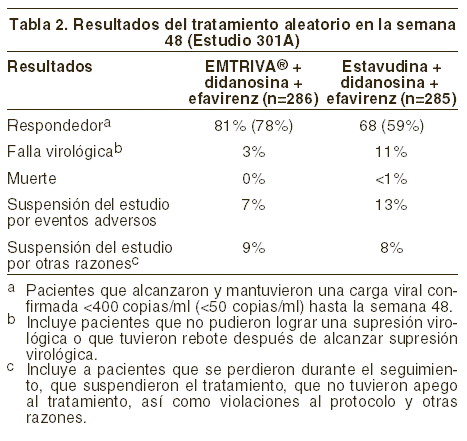
El incremento medio en el recuento celular CD+, con respecto al valor basal, fue de 168 células/mm3 en el grupo de EMTRIVA® y de 134 células/mm3 en el grupo de estavudina. En el transcurso de las 48 semanas en el grupo con EMTRIVA®, cinco pacientes (1,7%) experimentaron un nuevo evento Clase C del CDC, en comparación con siete pacientes (2,5%) del grupo de estavudina. Pacientes adultos que han recibido tratamiento previo: estudio 303: el estudio 303 fue un estudio abierto, multicéntrico, controlado con substancia activa, en el que se comparó EMTRIVA® (200 mg una vez al día), con lamivudina, en combinación con estavudina o zidovudina y un inhibidor de la proteasa o NNRTI en 440 pacientes que habían recibido doce semanas antes del estudio tratamiento antirretrovial triple, incluyendo lamivudina, y que tenían, al momento de ingresar al estudio, una carga viral ≤400 copias/ml. Los pacientes se seleccionaron de manera aleatoria 1:2 para continuar el tratamiento con lamivudina (150 mg dos veces al día) o bien para cambiar a EMTRIVA® (200 mg una vez al día). El resto del tratamiento se mantuvo sin cambios en todos los pacientes. La edad promedio de los sujetos fue de 42 años (intervalo 22-80), 86% fueron hombres; 64% caucásicos, 21% afroamericanos y 13% hispanos. Los pacientes tuvieron un recuento celular CD4+ basal medio de 527 células/mm3 (intervalo 37-1909), y una mediana del ARN del VIH-1 plasmático inicial de 1,7 log10 copias/ml (intervalo 1,7-4,0). La duración media del tratamiento antirretroviral previo fue de 27,6 meses. En la tabla 3, a continuación, se presentan los resultados del tratamiento.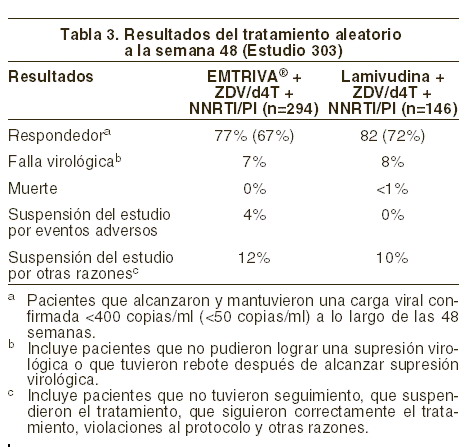
El incremento medio del conteo celular de células CD4 fue de 29 células/mm3 en el grupo de EMTRIVA® y 61 células/mm3 en el grupo de lamivudina. Pacientes pediátricos: en tres estudios clínicos abiertos y sin asignación aleatoria, se administró emtricitabina a 169 pacientes infectados por VIH-1, tanto naïve como con tratamiento antirretroviral previo (definidos como en supresión virológica recibiendo un esquema que contenía lamivudina en que este medicamento fue reemplazado por la emtricitabina), con edades comprendidas entre los tres meses y los 21 años. Los pacientes recibieron EMTRIVA® en solución oral, una vez al día (6 mg/kg, hasta un máximo de 240 mg/día) o EMTRIVA® en cápsulas (una sola cápsula de 200 mg una vez al día), asociado por lo menos a otros dos antirretrovirales. Los pacientes tenían una edad promedio de 7,9 años (intervalo, 0,3 a 21 años); el 49% eran varones, el 15% eran de raza blanca, el 61%, negros y el 24%, latinos. Los pacientes tuvieron una mediana del ARN del VIH-1 inicial de 4,6 log10 copias/ml (intervalo, 1,7 a 6,4), y un recuento promedio inicial de linfocitos CD4+ de 745 linfocitos/mm3 (intervalo, 2 a 2650). A las 48 semanas de tratamiento, la proporción total de pacientes que lograron y mantuvieron el ARN del VIH-1 < 400 copias/ml fue del 86%, y de < 50 copias/ml, del 73%. El aumento promedio del recuento de linfocitos CD4+ con respecto al valor inicial fue de 232 linfocitos/mm3 (-945, +1512). El perfil de reacciones adversas observadas en estos ensayos clínicos fue similar al de los pacientes adultos, salvo la aparición de anemia y la frecuencia más alta de hiperpigmentación en los niños (véase Reacciones secundarias y adversas). Se estudiaron las propiedades farmacocinéticas de la emtricitabina en 20 recién nacidos, nacidos de madres positivas para el VIH-1. Cada madre recibió tratamiento antirretroviral combinado antes del parto y durante éste. Los recién nacidos recibieron un tratamiento profiláctico con zidovudina durante un tiempo de hasta seis semanas después del nacimiento. Se administraron a los recién nacidos dos ciclos breves de emtricitabina en solución oral (cada uno, 3 m/kg. Una vez al día, durante cuatro días), durante los tres primeros meses de vida. Las exposiciones a la emtricitabina en los recién nacidos fueron similares a las exposiciones alcanzadas en los pacientes > 3 meses a 17 años (véase Farmacocinética y farmacodinamia: Poblaciones especiales, Pediatría). Durante los dos períodos cortos de administración de emtricitabina, en los recién nacidos tratados no se identificó ningún problema de toxicidad del fármaco. Todos los recién nacidos fueron negativos para el VIH-1 al final del estudio; no se pudo determinar la eficacia de la emtricitabina en la prevención o el tratamiento del VIH-1.
Farmacocinética y farmacodinamia: Descripción: EMTRIVA® es el nombre comercial de la emtricitabina, inhibidor de la transcriptasa reversa análogo de nucleósido (ITRAN) sintético con actividad contra el virus de la inmunodeficiencia humana tipo 1 (VIH-1). El nombre químico de emtricitabina es 5-fluoro-1 (2R, 5S)-[2 (hidroximetil)-1,3-oxatiolan-5-yl]citosina. La emtricitabina es el enantiómero (-) de un análogo tío de la citidina, diferente de otros análogos de la citidina porque tiene un flúor en la posición 5. Su fórmula molecular es C8H10FN3O2S y tiene un peso molecular de 247,24. Su fórmula estructural es la siguiente: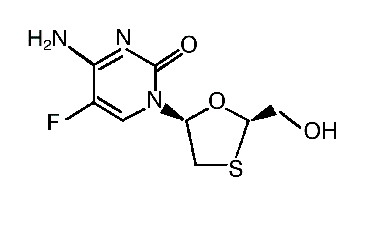
Emtricitabina es un polvo de blanco a blancuzco, con una solubilidad de aproximadamente 112 mg/ml en agua a 25°C. El logaritmo P de emtricitabina es -0,43 y el pKa es de 2,55. Las células de EMTRIVA® son para administración oral. Cada cápsula contiene 200 mg de emtricitabina; y sus ingredientes inactivos son crospovidona, estearato de magnesio, celulosa microcristalina y povidona. Mecanismo de acción: la emtricitabina es un antiviral. La emtricitabina, análogo nucleósido sintético de la citosina, es fosforilada por las enzimas celulares para obtener 5'-trifosfato de emtricitabina. Este compuesto inhibe la actividad de la transcriptasa reversa del VIH-1 al competir con el sustrato natural 5'trifosfato deoxicitidina y al incorporarse al ADN viral en formación, lo que produce la terminación de la cadena. El 5'trifosfato de emtricitabina es un inhibidor débil de las polimerasas de ADN a, b, e y de la polimerasa de ADN mitocondrial c de los humanos. Actividad antiviral: se evaluó la actividad antiviral de la emtricitabina en cultivo de células contra los aislados de laboratorio y clínicos de VIH en líneas de células linfoblastoides, en la línea celular MAGI-CCR5 y en células mononucleares de sangre periférica. El valor de concentración efectiva al 50% (EC50) de emtricitabina estuvo en el intervalo de 0,0013 y 0,64 mM (0,0003 a 0,158 mg/ml). Los estudios sobre la combinación de emtricitabina con los inhibidores de la transcriptasa reversa nucleósidos (abacavir, lamivudina, estavudina, tenofovir, zalcitabina, zidovudina), los inhibidores de la transcriptasa inversa no nucleósidos (delavirdina, efavirenz, nevirapina) y los inhibidores de la proteasa (amprenavir, nelfinavir, ritonavir, saquinavir) mostraron efectos de aditivos a sinérgicos. Emtricitabina presentó una actividad antiviral en cultivo de células contra las clases A, C, D, E, F y G del VIH-1 (los valores IC50 oscilaron de 0,007 a 0,075 mM) y mostraron actividad específica contra el VIH-2 (los valores IC50 oscilaron de 0,007 a 1,5 mM). En dos estudios clínicos se evaluó la actividad in vivo de emtricitabina, en los cuales a 101 pacientes se les administró de 25-40 mg de EMTRIVA® al día como monoterapia, de 10-14 días. Se observó un efecto antiviral referente a la dosis, con una disminución media de la Carga Viral de 1,3 log10 con la dosis de 200 mg una vez o dos veces al día. Resistencia: se ha logrado aislar, en cultivos de células e in vivo, cepas de VIH-1 resistentes a emtricitabina. El análisis genotípico de estos aislados mostró que la susceptibilidad reducida a emtricitabina estaba asociada con una mutación en el cordón 184 del gen de la transcriptasa reversa del VIH-1, la cual dio por resultado una situación del aminoácido metionina por valina o isoleucina (M184V/I). Se han recuperado cepas de VIH-1 resistentes a emtricitabina de pacientes ya tratados con este fármaco, solo o en combinación con otros agentes antirretrovirales. En un estudio clínico de pacientes vírgenes al tratamiento tratados con EMTRIVA, didanosina y efavirenz (véase Indicaciones terapéuticas, Estudios clínicos) los aislados virales de 37,5% de los pacientes que presentaron falla virológica mostraron susceptibilidad reducida a la emtricitabina. El análisis genotípico de estos aislados demostró que la resistencia se debió a M184V/I en el gen de la transcriptasa reversa de VIH-1. En un estudio clínico de pacientes sin tratamiento previo con antirretrovirales, tratados con EMTRIVA®, VIREAD® y efavirenz, o con zidovudina, lamivudina y efavirenz (véase Indicaciones terapéuticas, Estudios clínicos), se realizaron análisis de resistencia en las cepas aisladas del VIH-1 de todos los pacientes con confirmación del fracaso virológico y con > 400 copias/ml de ARN del VIH-1 en la 144ª semana o que interrumpieron precozmente el tratamiento. La aparición de sustituciones asociadas a la resistencia al efavirenz se produjo con mayor frecuencia y fue similar en todos los grupos de tratamiento. Se observó la sustitución de aminoácidos M184V, asociada a la resistencia a EMTRIVA® y a la lamivudina, en 2/19 cepas aisladas de pacientes analizados en el grupo tratado con zidovudina y lamivudina. En las 144 semanas del estudio 934, ningún paciente había presentado una sustitución K65R detectables en su VIH-1, según los análisis genotípicos habituales. Resistencia cruzada: se encontró que existe resistencia cruzada entre ciertos medicamentos del grupo de los ITRAN. Las cepas del VIH resistentes a emtricitabina (M184V/I) mostraron resistencia cruzada a lamivudina y zalcitabina, pero conservaron sensibilidad en cultivo de células a la didanosina, la estavudina, el tenofovir, la zidovudina y a los NNRTI (delavirdina, efavirenz y nevirapina). Las cepas del VIH-1 que contienen la sustitución K65R, seleccionada in vivo por abacavir, didanosina, tenofovir y zalcitabina mostraron una susceptibilidad reducida a la inhibición por emtricitabina. Los virus que albergan sustituciones que confieren susceptibilidad reducida a la estavudina y a la zidovudina (M41L, D67N, K70R, L210W, T215Y/F, K219Q/E) o a la didanosina (L74V) conservaron sensibilidad a emtricitabina. El VIH-1 que contiene la sustitución K103N, asociada con resistencia a los NNRTI fue susceptible a la emtricitabina. Farmacocinética en adultos: la farmacocinética de emtricitabina se evaluó en voluntarios sanos y en individuos infectados con VIH-1. Las farmacocinéticas de la emtricitabina son similares entre estas poblaciones. En la figura 1 se muestra la curva tiempo-concentración plasmática media en estado estable de la emtricitabina, en 20 sujetos infectados con el VIH-1 que recibieron EMTRIVA® Cápsulas.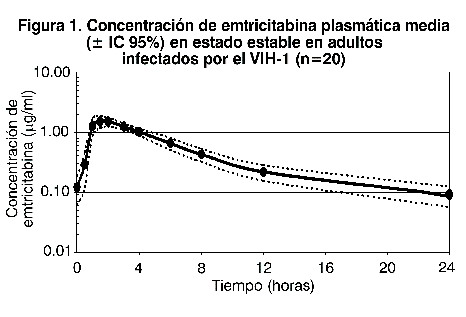
Absorción: la emtricitabina se absorbe extensa y rápidamente después de su administración oral, la concentración máxima en plasma se alcanza de una a dos horas después de la ingesta. Después de la administración oral de dosis múltiple de EMTRIVA® Cápsulas a 20 sujetos infectados con VIH-1, la concentración plasmática máxima (Cmáx) de emtricitabina en estado estable (media ± SD) fue de 1,8 = 0,7 mg/ml, y el área bajo la curva de tiempo de concentración plasmática a lo largo de un intervalo de dosificación de 24 horas (ABC) fue de 10,0 ± 3,1 hr*mg/ml. La concentración media en estado estable a las 24 horas posteriores a la dosis fue de 0,09 mg/ml. La biodisponibilidad absoluta media de EMTRIVA® fue de 93%. Los parámetros farmacocinéticos de la emtricitabina administrada en dosis múltiple son proporcionales a la dosis, en un rango entre 25 y 200 mg. Distribución: la liga de emtricitabina con las proteínas plasmáticas in vitro fue de < 4% e independiente de la concentración sobre el intervalo de 0,02-200 mg/ml. En la concentración plasmática máxima del medicamento, la proporción plasmática media en la sangre fue de <126>1,0 y la proporción media de concentración semen/plasma, de <126>4,0. Metabolismo: los estudios in vitro indican que la emtricitabina no es un inhibidor de la enzima humana CYP450. Después de la administración de emtricitabina-C14, se logró una recuperación completa de la dosis en la orina (<126>86%) y en las heces (<126>14%). Trece por ciento (13%) de la dosis se recuperó en la orina en forma de tres metabolitos. La biotransformación de la emtricitabina incluye la oxidación del tiol proporcional para formar los diastereómeros de 3-sulfóxido (<126>9% de la dosis y la conjugación con el ácido glucurónido para formar la 2'-O-glucuronida (<126>4% de la dosis). No se identificaron otros metabolitos. Eliminación: la vida media plasmática de la emtricitabina es aproximadamente de 10 horas. La depuración renal de emtricitabina es mayor que la depuración estimada de creatinina, lo que sugiere que la eliminación se da por una combinación entre filtración glomerular y secreción tubular activa. Puede existir competencia para la eliminación con otros compuestos que también son eliminados renalmente. Efectos de los alimentos en la absorción oral: la EMTRIVA® Cápsulas se puede tomar con o sin alimentos. Al administrar EMTRIVA® Cápsulas junto con alimentos (comida alta en grasas de aproximadamente 1.000 kcal), los valores de exposición sistémica de emtricitabina (ABC) no sufrieron alteración alguna, mientras que la Cmáx disminuyó en 29%). Poblaciones especiales: raza, sexo: las farmacocinéticas de la emtricitabina fueron similares en pacientes hombres y mujeres adultos; tampoco se encontraron diferencias farmacocinéticas atribuibles a la raza. Pediatría: la farmacocinética de emtricitabina en estado estable fue determinada en 77 niños infectados con VIH-1, y se caracterizó el perfil de farmacocinética en cuatro grupos etarios (tabla 4). La exposición a emtricitabina obtenida en niños que recibieron una dosis diaria de 6 mg/kg hasta un máximo de 240 mg en forma de solución oral o una cápsula de 200 mg es similar a las exposiciones obtenidas en adultos que recibieron una dosis de una vez al día de 200 mg. Se estudió la farmacocinética de emtricitabina en 20 neonatos nacidos de madres VIH-1 positivas. Cada madre recibió prenatal e intraparto una terapia antirretroviral de combinación. A modo profiláctico, los neonatos recibieron zidovudina hasta 6 semanas después del nacimiento. Se administró a los neonatos dos tratamientos cortos de emtricitabina en forma de solución oral (cada uno 3 mg/kg una vez al día x 4 días) durante los primeros 3 meses de vida. El área bajo la curva (ABC) observada en neonatos que recibieron una dosis diaria de 3 mg/kg de emtricitabina fue similar al ABC observada en pacientes pediátricos ≥3 meses a 17 años de edad que recibieron una dosis diaria de emtricitabina en forma de solución oral de 6 mg/kg hasta 240 mg o como una cápsula de 200 mg (tabla 4).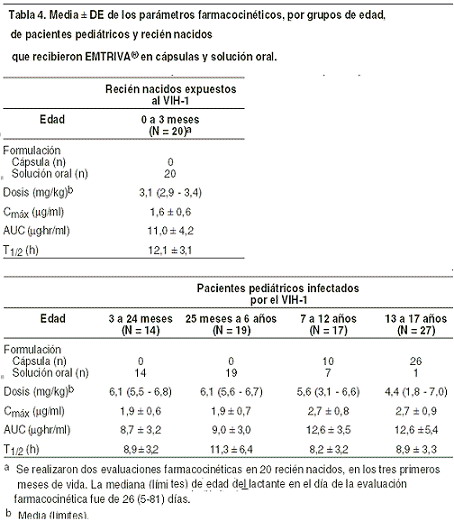
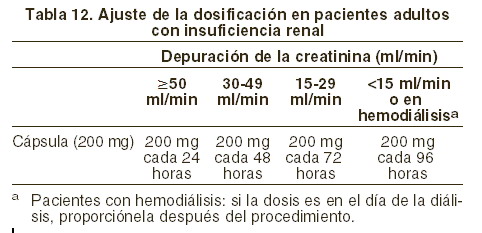
Pacientes geriátricos: la farmacocinética de emtricitabina no se ha evaluado completamente en personas mayores. Pacientes con disfunción renal: la farmacocinética de emtricitabina se altera en pacientes con insuficiencia renal (Precauciones generales). En pacientes con depuración de creatinina < 50 ml/min o con insuficiencia renal terminal (IRT) que requiere diálisis, la Cmáx y la ABC de emtricitabina aumentaron debido a la reducción en la depuración renal (tabla 5). Se recomienda que el intervalo de dosificación de EMTRIVA® se modifique en pacientes con depuración de creatinina < 50 ml/min o en pacientes con IRT que requieran diálisis (véase Dosis y vía de administración). En pacientes pediátricos, se desconocen los efectos del daño renal en la farmacocinética de emtricitabina.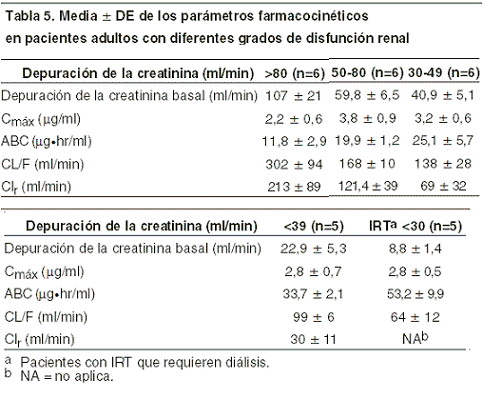
Hemodiálisis: un tratamiento hemodiálico elimina aproximadamente 30% de la dosis de emtricitabina a lo largo de una diálisis de 3 horas; iniciado 1,5 horas después de la dosificación, permite eliminar (una proporción de flujo sanguíneo de 400 ml/min y en proporción de flujo dializado de 600ml/min). Se desconoce si la emtricitabina se puede eliminar por diálisis peritoneal. Pacientes con disfunción hepática: tampoco se ha estudiado en pacientes con daño hepático; sin embargo, como la emtricitabina no es metabolizada por las enzimas del hígado, el impacto de una hepatopatía debe ser mínimo.
Contraindicaciones: El EMTRIVA® está contraindicado para pacientes con hipersensibilidad previamente demostrada a cualquiera de los componentes de los productos.
Precauciones generales: Acidosis láctica/hepatomegalia severa con esteatosis: se han reportado casos de acidosis láctica y hepatomegalia severa con esteatosis, incluyendo casos fatales asociados con el uso de análogos nucleósidos (ITRAN) solos o en combinación, incluyendo emtricitabina y otros antirretrovirales. La mayoría de estos casos se han presentado en mujeres. La obesidad y la exposición prolongada a los nucleósidos parecen ser factores de riesgo. Se debe tener especial precaución cuando se administran análogos nucleósidos a cualquier paciente con factores de riesgo conocidos para enfermedades hepáticas. Sin embargo, también se han reportado casos en pacientes sin estas características. El tratamiento con EMTRIVA® deberá suspenderse en cualquier paciente que en estudios clínicos o de laboratorio se le descubran aspectos que sugieran acidosis láctica o hepatotoxicidad pronunciada (la cual puede incluir hepatomegalia y esteatosis aún en ausencia de hipertransaminasemia pronunciada). Pacientes coinfectados con VIH-1 y el virus de hepatitis B: se recomienda que, antes de iniciar el tratamiento antirretroviral, se haga una prueba a todos los pacientes con VIH-1 para ver si tienen presente el virus de hepatitis B (VHB) crónico. EMTRIVA® no ha sido aprobado para el tratamiento de la infección crónica por VHB, no se han establecido la seguridad y la eficacia de EMTRIVA® en pacientes coinfectados con VHB y VIH-1. Se han reportado exacerbaciones agudas severas de hepatitis B en pacientes después de haber discontinuado EMTRIVA®. En algunos pacientes infectados por el VHB y tratados con EMTRIVA®, las exacerbaciones de hepatitis B se asociaron con descompensación hepática e insuficiencia hepática. La función hepática debería ser monitoreada minuciosamente, tanto por seguimiento clínico como de laboratorio, por lo menos durante varios meses en pacientes que estén coinfectados con VIH-1 y VHB y que discontinúen EMTRIVA®. Si es apropiado, puede justificarse la iniciación del tratamiento anti-hepatitis B. Administración concomitante de productos relacionados: EMTRIVA® es un componente de TRUVADA® (una asociación de dosis fija de emtricitabina y tenofovir disoproxil fumarato) y de ATRIPLATM (una asociación de dosis fija de efavirenz, emtricitabina y tenofovir disoproxil fumarato). EMTRIVA® no se debe administrar concomitantemente con TRUVADA® ni con ATRIPLATM. Debido a las semejanzas entre la emtricitabina y la lamivudina, no se debe administrar EMTRIVA® concomitantemente con otros fármacos que contengan lamivudina, como Combivir™ (lamivudina y zidovudina), Epivir™ o Epivir-VHB™ (lamivudina), Epzicom™ (sulfato de abacavir y lamivudina) o Trizivir™ (sulfato de abacavir, lamivudina y zidovudina). Nueva aparición o empeoramiento de la disfunción renal: la emtricitabina se elimina principalmente por el riñón. Se recomienda la reducción de la dosis de EMTRIVA® para pacientes con disfunción renal. Se recomienda modificar la dosis o el intervalo de dosificación de EMTRIVA® en los pacientes con una depuración de creatinina < 50 ml/min o en los pacientes que precisan diálisis (véase Farmacocinética y farmacodinamia, Poblaciones especiales; Dosis y vía de administración). Redistribución de la grasa corporal: en pacientes tratados con antirretroviral, se han observado eventos de redistribución/acumulación de grasa corporal, incluyendo obesidad central, hiperplasia de grasa dorso-cervical (joroba de búfalo), emaciación periférica o emaciación facial, agrandamiento de mamas y "aspecto cushinoide". Actualmente, se desconoce el mecanismo y las consecuencias a largo plazo de estos eventos. No se ha establecido una relación causal. Síndrome de reconstitución inmune: se ha reportado el síndrome de reconstitución inmune en pacientes tratados con terapia antirretroviral de combinación, incluyendo EMTRIVA®. Durante la fase inicial del tratamiento antirretroviral de combinación, los pacientes cuyo sistema inmune responde pueden desarrollar una respuesta inflamatoria a infecciones oportunistas indolentes o residuales (como la infección por Mycobacterium avium, citomegalovirus, Pneumocystis jirovecii pneumonia (PCP) o tuberculosis), que podrían requerir de una evaluación más a fondo y adicional. Uso pediátrico: la seguridad y la eficacia de la emtricitabina en los pacientes de tres meses a 21 años de edad son apoyados por los datos obtenidos de tres estudios clínicos abiertos y no aleatorizados, en que se administró emtricitabina a 169 pacientes infectados por el VIH-1, tanto naïve como con tratamiento antirretroviral previo (definidos como en supresión virológica bajo un esquema que contenía lamivudina en que este medicamento fue reemplazado por la emtricitabina) (véase Indicaciones terapéuticas, Estudios clínicos). Se estudiaron las propiedades farmacocinéticas de la emtricitabina en 20 recién nacidos, nacidos de madres positivas para el VIH-1 (véase Indicaciones terapéuticas, Estudios clínicos). Todos los recién nacidos fueron negativos para el VIH-1 al final del estudio; no se pudo determinar la eficacia de la emtricitabina en la prevención o el tratamiento del VIH-1. Uso geriátrico: los estudios clínicos de EMTRIVA® no incluyeron el número suficiente de pacientes de 65 años y mayores, a fin de determinar si éstos responden de manera diferente de las personas más jóvenes. En general, la selección de dosis para el paciente mayor debería ser cautelosa, teniendo en mente la mayor frecuencia de padecimientos hepáticos, renales o cardíacos, y de enfermedad concomitante u otros tratamientos farmacológicos. Información para el paciente: los pacientes deben ser informados de que: el EMTRIVA® no cura la infección por el VIH-1; los pacientes que lo tomen pueden seguir experimentando enfermedades relacionadas con la infección, incluyendo infecciones oportunistas. Todo paciente bajo tratamiento de EMTRIVA® deberá estar bajo la supervisión de un médico. No se ha demostrado que el uso de EMTRIVA® reduzca el riesgo de transmisión del VIH-1 a otros mediante el contacto sexual o transfusión sanguínea. Se desconocen los efectos a largo plazo de EMTRIVA®. Las cápsulas de EMTRIVA® son exclusivamente para administración oral. Al igual que con otros antirretrovirales, es fundamental seguir el tratamiento con EMTRIVA® al pie de la letra. Se ha notificado la aparición de acidosis láctica y hepatomegalia grave con esteatosis, incluso casos mortales. El tratamiento con EMTRIVA® debe interrumpirse en cualquier paciente que presente síntomas clínicos que indiquen la presencia de acidosis láctica o de hepatotoxicidad pronunciada (incluso náuseas, vómitos, molestias gástricas poco habituales o inesperadas y debilidad) (véase Precauciones generales). Se debe realizar a los pacientes con infección por el VIH-1 la prueba para detectar la presencia del virus de la hepatitis B (VIH) antes de iniciar el tratamiento antirretroviral. Se han notificado casos de exacerbación aguda y grave de la hepatitis B en los pacientes infectados concomitantemente por el VHB y el VIH-1 que han interrumpido la administración de EMTRIVA®. EMTRIVA® no debe administrarse concomitantemente con otros fármacos que contengan lamivudina, como Combivir™ (lamivudina y zidovudina), Epivir™ o Epivir™-HBV (lamivudina), Epzicon™ (sulfato de abacavir y lamivudina) o Trizivir™ (sulfato de abacavir, lamivudina y zidovudina) (véase Precauciones generales). Puede ser necesario ajustar la dosis o el intervalo de dosificación de EMTRIVA® en los pacientes con disfunción renal (véase Dosis y vía de administración).
Restricciones de uso durante el embarazo y la lactancia: Embarazo: embarazo categoría B: los estudios de toxicidad embriofetal realizados en ratones con concentraciones (ABS) hasta 60 veces mayores, y en conejos, con concentraciones hasta 120 veces por arriba de la exposición en humanos con la dosis diaria recomendada, no revelaron una mayor incidencia de variaciones o malformaciones fetales. Sin embargo, no existen estudios adecuados y bien controlados en mujeres embarazadas. Dado que los estudios sobre reproducción en modelos animales no siempre permiten predecir lo que ocurrirá en humanos, el EMTRIVA® deberá utilizarse durante el embarazo sólo si es totalmente necesario. Madres lactando: los centros de control y prevención de enfermedades recomiendan que las madres infectadas con VIH no den pecho a sus hijos para evitar el riesgo de transmisión postnatal del VIH. Se desconoce si la emtricitabina es excretada en la leche materna humana. Debido tanto al riesgo de transmisión del VIH como al potencial de reacciones adversas serias que puede haber en los lactantes, se debe pedir a las madres que no den pecho a sus bebes si están tomando EMTRIVA®.
Reacciones secundarias y adversas: En otros apartados de la información para prescribir se tratan las siguientes reacciones adversas: acidosis láctica o hepatomegalia grave con esteatosis (véase Precauciones generales). Exacerbaciones agudas y graves de la hepatitis B (véase Precauciones generales). Síndrome de reconstitución inmunológica (véase Precauciones generales). Reacciones adversas de la experiencia en ensayos clínicos: pacientes adultos: más de 2.000 pacientes adultos con infección por VIH han sido tratados con EMTRIVA® solo o en combinación con otros agentes antirretrovirales durante períodos que van de 10 días a 200 semanas en estudios clínicos, Fases I-III. Debido a que los ensayos clínicos se realizan en condiciones muy variadas, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden compararse directamente con las tasas en los ensayos clínicos de otro fármaco y puede que no reflejen las tasas observadas en la práctica. Las reacciones adversas más frecuentes (incidencia ≥10%, cualquier gravedad) identificadas a partir de cualquiera de los tres grandes ensayos clínicos controlados son cefalea, diarrea, náuseas, fatiga, mareos, depresión, insomnio, anomalías de los sueños, erupción cutánea, dolor abdominal, astenia, aumento de la tos y rinitis. Estudios 301A y 303: reacciones adversas aparecidas con el tratamiento: las reacciones adversas más frecuentes que se produjeron en los pacientes que recibieron EMTRIVA® con otros antirretrovirales en los estudios 301 y 303 fueron: cefalea, diarrea, náusea y erupción cutánea, cuya severidad por lo general fue de leve a moderada. Aproximadamente 1% de los pacientes discontinuó su participación en los estudios clínicos debido a estos eventos. Todas las reacciones adversas se reportaron con frecuencia similar en los grupos con EMTRIVA® y en los tratamientos de control, con la excepción de la decoloración en la piel la cual se presentó con mayor frecuencia en el grupo tratado con EMTRIVA®. La decoloración en la piel, manifestada como hiperpigmentación en las palmas y/o en las plantas de los pies, fue generalmente leve y asintomática. Se desconoce tanto su mecanismo como su importancia clínica. En la Tabla 6 se proporciona un resumen de las reacciones adversas clínicas registradas durante el tratamiento con EMTRIVA® de acuerdo con los estudios 301A y 303.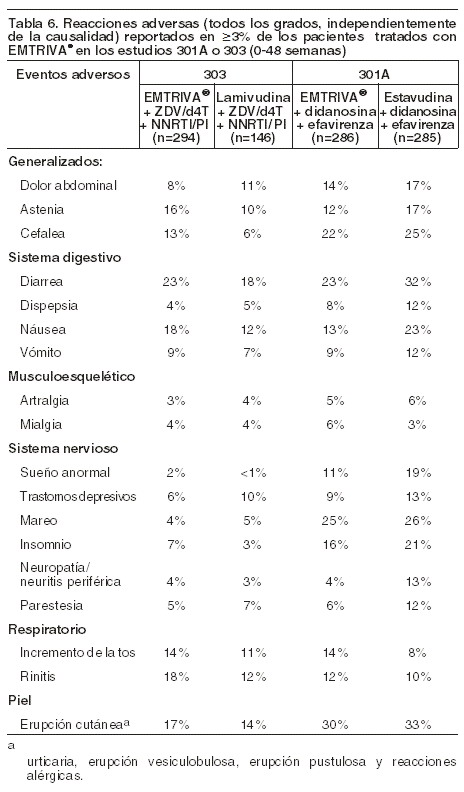
Estudio 934: reacciones adversas aparecidas con el tratamiento: en el estudio 934, se administró a 511 pacientes sin tratamiento antirretroviral previo VIREAD® + EMTRIVA® asociado a efavirenz (N=257), o bien zidovudina y lamivudina asociados a efavirenz (N=254). Las reacciones adversas observadas en este estudio fueron en general consistentes con las observadas en estudios anteriores en los pacientes con o sin tratamiento previo con antirretrovirales (tabla 7):
Pacientes pediátricos: la evaluación de las reacciones adversas se basa en los datos del estudio 203, un estudio abierto y no controlado, de 116 pacientes pediátricos infectados por el VIH-1 que recibieron emtricitabina durante 48 semanas. En general, el perfil de reacciones adversas en los pacientes pediátricos fue comparable al observado en los estudios clínicos de EMTRIVA® en los pacientes adultos. La hiperpigmentación fue más frecuente en los niños. Entre las reacciones adversas adicionales que se identificaron en este estudio se cuenta la anemia. Algunos acontecimientos adversos que aparecieron con el tratamiento, con independencia de la causalidad, notificados en los pacientes durante 48 semanas de tratamiento fueron los siguientes: infección (44%), hiperpigmentación (32%), aumento de la tos (28%), vómitos (23%), otitis media (23%), erupción cutánea (21%), rinitis (29%), diarrea (20%), fiebre (18%), neumonía (15%), gastroenteritis (11%), dolor abdominal (10%) y anemia (7%). Las anomalías de laboratorio de 3° y 4° grados, que aparecieron con el tratamiento, las sufrió el 9% de los pacientes pediátricos, y consistieron en amilasa > 2,0 x LSN (n=4), neutrófilos < 750/mm3 (n=3), ALT > 5 x LSN (n=2), aumento de la CPK ( > 4 x LSN)(n=2) y un paciente cada uno con aumento de la bilirrubina ( > 3,0 x LSN), aumento de la GGT ( > 10 x LSN), aumento de la lipasa ( > 2,5 x LSN), disminución de la hemoglobina ( < 7 g/dl), y disminución de la glucosa ( < 40 mg/dl).
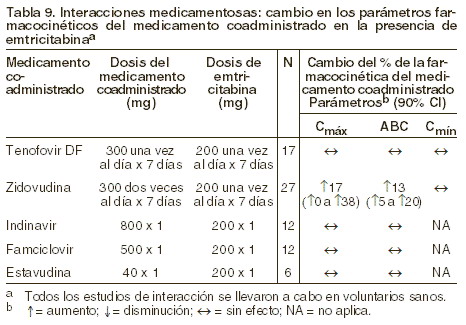
Interacciones medicamentosas y de otro género: Se ha estudiado la posibilidad de interacciones medicamentosas con EMTRIVA® asociado a zidovudina, indinavir, estavudina, famciclovir y tenofovir disoproxil fumarato. No se observaron interacciones medicamentosas clínicamente significativas con ninguno de estos fármacos. Evaluación de las interacciones medicamentosas: en concentraciones hasta 14 veces más altas que las observadas in vivo, la emtricitabina no inhibió in vitro al metabolismo de medicamentos por alguna de las siguientes isoformas humanas de CYP: CYP1A2, CYP2B6, CYP2C19, CYP2D6 y CYP3A4. La emtricitabina tampoco inhibió a la enzima responsable de la glucoronidación (uridina-5´-disfosfoglucoronil transferasa). Basándose en los resultados de estos experimentos in vitro y en la ruta conocida de eliminación de la emtricitabina (renal), puede concluirse que bajó el potencial del CYP mediante interacciones que incluyen emtricitabina con otros medicamentos. EMTRIVA® se evaluó en voluntarios sanos en combinación con tenofovir disoproxil fumarato (TDF), zidovudina, indinavir, famciclovir y estavudina. Las tablas 8 y 9 resumen los efectos farmacocinéticos del medicamento coadministrado a la farmacocinética de emtricitabina en los efectos de emtricitabina sobre la farmacocinética del fármaco coadministrado.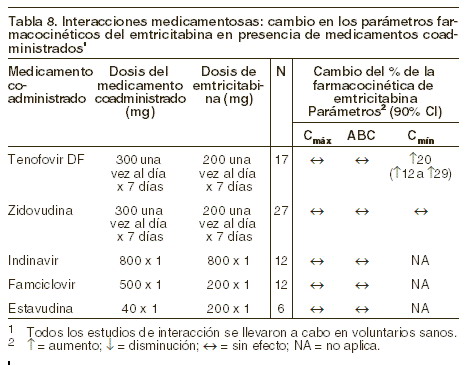
Alteraciones en los resultados de pruebas de laboratorio: Estudios 301A y 303: durante los estudios clínicos ya mencionados, las alteraciones de laboratorio ocurrieron con frecuencia similares en los grupos tratados con EMTRIVA® y en los grupos control. En la tabla 10 se proporciona un resumen de las anormalidades de laboratorio grados 3 y 4.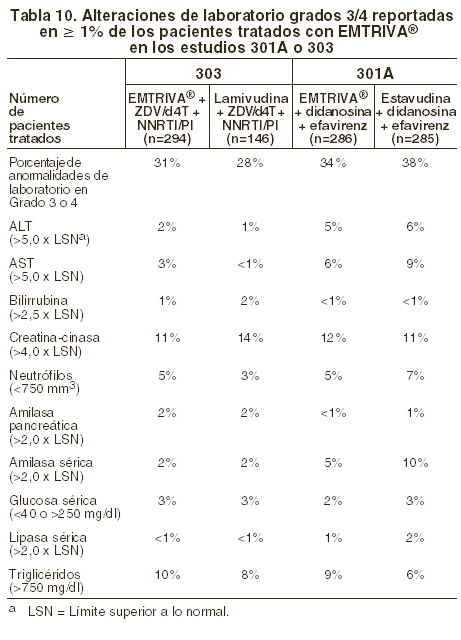
Estudio 934: en la tabla 11 se muestran las anormalidades de valores de laboratorio significativas que se observaron en este estudio.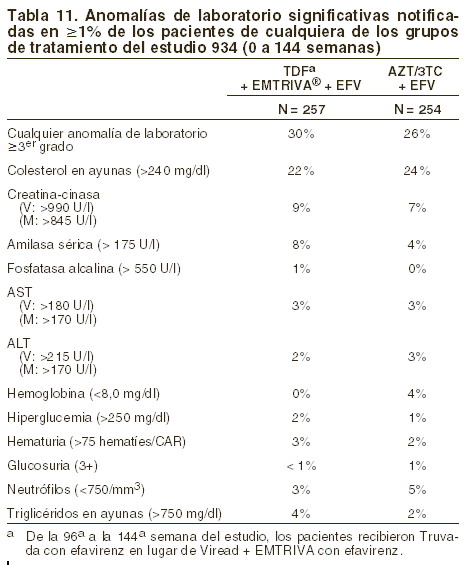
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: En estudios de carcinogénesis oral a largo plazo con emtricitabina, no hubo aumentos de incidencia de tumor relacionados con fármaco en ratones a dosis de hasta 750 mg/kg/día (26 veces la exposición sistémica humana a la dosis terapéutica de 200 mg/día) o en ratas a dosis de hasta 600 mg/kg/día (31 veces la exposición sistémica humana a la dosis terapéutica). En la prueba bacterial de mutación reversa (prueba de Ames), el EMTRIVA® no fue genotóxico en las pruebas en linfoma de ratón y en micronúcleos de ratón. La emtricitabina no afectó la fertilidad de ratas machos sometidos a concentraciones 140 veces mayores, o de ratones machos y hembras sometidos a exposiciones 60 veces mayores (ABC) a las de humanos adultos, a los que se les dio la dosis diaria recomendada de 200 mg. La fertilidad fue normal en las crías de ratones expuestos diariamente, desde antes de su nacimiento (en el útero) y hasta su madurez sexual, a exposiciones diarias (ABC) aproximadamente 60 veces más altas que las exposiciones de seres humanos a la dosis diaria recomendada de 200 mg.
Dosis y vía de administración: Dosis recomendada: EMTRIVA® puede tomarse independientemente de los alimentos. Pacientes adultos (18 años de edad y mayores): EMTRIVA® Cápsulas: una cápsula de 200 mg administrada una vez al día por vía oral. Niños y adolescentes hasta 18 años de edad: EMTRIVA® Cápsulas: para niños y adolescentes que pesen más de 33 kg que puedan tragar una cápsula intacta, una cápsula de 200 mg administrada una vez al día por vía oral.