EPIVAL SPRINKLE
ABBOTT
Denominación genérica: Valproico, Ácido (valproato)
Forma farmacéutica y formulación: Cápsulas. Cada cápsula de EPIVAL® SPRINKLE contiene: Valproato semisódico equivalente a 125 mg de ácido valproico; Excipiente, c.b.p. 1 cápsula Sprinkle.
Indicaciones terapéuticas: Anticonvulsivante. Epilepsia: EPIVAL® SPRINKLE está indicado como monoterapia y terapia complementaria en el tratamiento de pacientes con crisis parciales complejas que ocurren en forma aislada o asociadas con otro tipo de crisis. EPIVAL® SPRINKLE también está indicado en monoterapia o terapia complementaria para el tratamiento de las crisis de ausencia simple y compleja y de manera complementaria en pacientes con múltiples tipos de crisis que incluyen crisis de ausencias. Se define ausencia simple como una muy breve pérdida del sensorio o de conciencia acompañada por ciertas descargas epilépticas generalizadas sin otros signos clínicos detectables. Ausencia compleja es el término usado cuando otros signos también están presentes. Ver Precauciones generales para la declaración respecto a la disfunción hepática fatal.
Farmacocinética y farmacodinamia: Farmacodinamia: El valproato semisódico se disocia a ion valproato en el tracto gastrointestinal. El mecanismo de acción por el cual el valproato ejerce su efecto terapéutico no ha sido del todo aclarado. Se sugiere que su actividad en epilepsia está relacionada con un aumento de los niveles cerebrales de ácido gamaaminobutírico (GABA). Farmacocinética-absorción/biodisponibilidad: Dosis orales equivalentes de valproato semisódico y ácido valproico aportan cantidades equivalentes de ion valproato a nivel sistémico. Aunque la velocidad de absorción del ion valproato puede variar con la formulación administrada (líquido, sólido o sprinkle), con las condiciones de uso (v.gr. ayuno o posprandio), y el método de administración (ya sea que el contenido de la cápsula se rocíe sobre el alimento o se tome intacta la cápsula), estas diferencias deben ser de menor importancia clínica bajo condiciones en estado estable alcanzadas con el uso crónico en el tratamiento de epilepsia. Sin embargo, es posible que las diferencias entre los productos de valproato en Tmax y Cmax pudieran ser importantes al inicio del tratamiento. Por ejemplo, en estudios de una sola dosis, el efecto del alimento tuvo una mayor influencia en la velocidad de absorción de la tableta (incremento en Tmax de 4 a 8 horas) que sobre la absorción de las cápsulas sprinkle (incrementó en Tmax de 3.3 a 4.8 horas). Mientras la velocidad de absorción desde el aparato G.I. y la fluctuación en las concentraciones plasmáticas de valproato varían con el régimen de dosificación y la formulación, la eficacia de valproato como un anticonvulsivo en el uso crónico es improbable que sea afectado. Con base en la experiencia, empleando dosificaciones de una vez al día a cuatro veces al día, así como en estudios en modelos de epilepsia en primates que involucró una velocidad de infusión constante, indica que la biodisponibilidad sistémica total (grado de absorción) es el principal determinante en el control de las crisis y que las diferencias en los cocientes de las concentraciones plasmáticas pico a valle entre las formulaciones de valproato son intrascendentes desde un punto de vista clínico práctico. La coadministración de productos orales de valproato con alimento y la sustitución entre varias formulaciones de valproato semisódico y ácido valproico no debe causar problemas clínicos en el manejo de pacientes con epilepsia (ver Dosis y vía de administración). No obstante, ante cualquier cambio en dosificación, o la adición o discontinuación de medicamentos concomitantes, debe ser cuidadosamente vigilado el estado clínico y las concentraciones plasmáticas de valproato.Farmacocinética-distribución: Unión a proteínas: La unión de valproato a proteínas plasmáticas depende de la concentración y la fracción libre aumenta aproximadamente de 10% con 40 mg/mL a 18.5% con 130 mg/mL. La unión a proteínas de valproato disminuye en los ancianos, en pacientes con enfermedades hepáticas crónicas, en pacientes con insuficiencia renal y en presencia de otros fármacos (por ejemplo, ácido acetilsalicílico). A la inversa, valproato puede desplazar ciertos fármacos unidos a proteínas (por ejemplo, fenitoína, carbamazepina, warfarina y tolbutamida) (ver Interacciones medicamentosas y de otro género). Distribución en el SNC: Las concentraciones de valproato en el líquido cefalorraquídeo (LCR) se aproximan a las concentraciones libres en plasma (alrededor de 10% de la concentración total). Farmacocinética-metabolismo: El valproato es metabolizado casi totalmente por el hígado. En pacientes adultos en monoterapia, de 30 a 50% de una dosis administrada aparece en orina como un conjugado glucurónido. La ß-oxidación mitocondrial es otra vía metabólica principal, representando normalmente más de 40% de la dosis. Por lo regular, menos de 15 a 20% de la dosis se elimina mediante otros mecanismos oxidativos. Menos de 3% de una dosis administrada se excreta intacta en orina. La relación entre la dosis y la concentración total de valproato es no lineal; la concentración no aumenta proporcionalmente con la dosis, más bien aumenta en menor grado debido a la unión saturable a proteínas plasmáticas. La cinética del fármaco libre es lineal. Farmacocinética-eliminación: El promedio de depuración plasmática y el volumen de distribución de valproato total son 0.56 L/h/1.73 m2 y 11 L/1.73 m2, respectivamente. El promedio de depuración plasmática y el volumen de distribución de valproato libre son 4.6 L/h/1.73 m2 y 92 L/1.73 m2, respectivamente. La vida media terminal promedio de la monoterapia con valproato varió de 9 a 16 horas después de los esquemas de dosificación oral de 250 a 1,000 mg. Los estimados citados corresponden principalmente a pacientes que no están tomando fármacos que afectan los sistemas de enzimas hepáticas metabólicas. Por ejemplo, los pacientes que toman fármacos antiepilépticos que inducen enzimas (carbamazepina, fenitoína y fenobarbital) eliminarán el valproato más rápidamente. Debido a estos cambios en la depuración de valproato, se debe intensificar el monitoreo de las concentraciones de antiepilépticos siempre que se introduzcan o retiren antiepilépticos concomitantes. Poblaciones especiales: Neonatos: Los niños en los primeros dos meses de vida tienen una capacidad marcadamente reducida para eliminar el valproato en comparación con niños de mayor edad y adultos. Esto es el resultado de una menor depuración (tal vez debido al retraso en el desarrollo de glucuronosiltransferasa y otros sistemas enzimáticos implicados en la eliminación de valproato) así como un mayor volumen de distribución (debido en parte a la menor unión a proteínas plasmáticas). Por ejemplo, en un estudio, la vida media en niños menores de 10 días varió de 10 a 67 horas en comparación con un rango de 7 a 13 horas en niños mayores de dos meses. Niños: Los pacientes pediátricos (es decir, entre 3 meses y 10 años) tienen depuraciones 50% más altas expresadas en peso (es decir, mL/min/kg) que los adultos. Los niños mayores de 10 años tienen parámetros farmacocinéticos que se aproximan a los de los adultos. Ancianos: Se ha demostrado que la capacidad de los pacientes ancianos (rango de edad: 68 a 89 años) para eliminar el valproato es reducida en comparación con la observada en adultos jóvenes (rango de edad: 22 a 26). La depuración intrínseca disminuye 39%; la fracción libre de valproato aumenta en 44%. Por consiguiente, en los ancianos se debe reducir la dosis inicial (ver Dosis y vía de administración). Sexo: No hay diferencias en el área de superficie corporal entre los varones y mujeres (4.8 ± 0.17 y 4.7 ± 0.07 L/h por 1.73 m2, respectivamente). Origen étnico: No se han estudiado los efectos del origen étnico en la cinética de valproato. Enfermedad hepática: Ver Contraindicaciones y Precauciones generales-Hepatotoxicidad. La enfermedad hepática deteriora la capacidad de eliminar el valproato. En un estudio, la depuración de valproato libre disminuyó en 50% en siete pacientes con cirrosis y en 16% en cuatro pacientes con hepatitis aguda, en comparación con seis personas sanas. En ese estudio, la vida media de valproato aumentó de 12 a 18 horas. La enfermedad hepática también se relaciona con concentraciones más bajas de albúmina y fracciones libres más grandes de valproato (aumentó de 2 a 2.6 veces). En consecuencia, el monitoreo de las concentraciones totales puede ser engañoso ya que las concentraciones libres pueden ser sustancialmente elevadas en pacientes con enfermedad hepática mientras que las concentraciones totales pueden parecer normales. Enfermedad renal: Se reportó una ligera reducción (27%) en la depuración de valproato libre en pacientes con insuficiencia renal (depuración de creatinina < 10 mL/min). Sin embargo, la hemodiálisis normalmente reduce las concentraciones de valproato en alrededor de 20%. Por consiguiente, no parece necesario ajustar la dosis en pacientes con insuficiencia renal. En estos pacientes la unión a proteínas disminuye sustancialmente; por consiguiente, el monitoreo de concentraciones totales puede ser engañoso. Niveles plasmáticos y efecto clínico: La relación entre la concentración plasmática y respuesta clínica no está bien documentada. Un factor que contribuye es la concentración no lineal dependiente de la unión a proteínas de valproato que afecta la depuración del fármaco. Por consiguiente, el monitoreo de valproato sérico total no puede proporcionar un índice confiable de las especies bioactivas de valproato. Por ejemplo, debido a que la unión de valproato a proteínas plasmáticas depende de la concentración, la fracción libre aumenta de aproximadamente 10% con 40 mg/mL a 18.5% con 130 mg/mL. En pacientes geriátricos, con hiperlipidemias o con enfermedades hepáticas y renales, se presentan fracciones libres más altas que lo esperado. Epilepsia: El rango terapéutico en epilepsia comúnmente se considera que es 50 a 100 mg/mL de valproato total, aunque algunos pacientes pueden ser controlados con concentraciones plasmáticas más bajas o más altas.
Contraindicaciones: El valproato semisódico no debe administrarse a pacientes con enfermedad hepática o disfunción hepática significativa (ver Precauciones generales-Hepatotoxicidad). El valproato semisódico está contraindicado en pacientes que tienen trastornos mitocondriales causados por mutaciones en la polimerasa g del DNA mitocondrial (POLG; v.gr. síndrome de Alpers-Huttenlocher) y en niños menores de dos años en quienes se sospecha tener un trastorno relacionado con POLG (ver Precauciones generales-Hepatotoxicidad). El valproato semisódico está contraindicado en pacientes con hipersensibilidad conocida al medicamento (ver Reacción de hipersensibilidad en múltiples órganos). El valproato semisódico está contraindicado en pacientes con trastorno en el ciclo de la urea (ver Precauciones generales-Trastorno del ciclo de la urea). El valproato semisódico está contraindicado en la profilaxis de migraña en mujeres embarazadas. El valproato semisódico está contraindicado en pacientes con porfiria.
Restricciones de uso durante el embarazo y la lactancia: Clasificación "O" de la FDA: Embarazo: Ver Contraindicaciones y Precauciones generales-Uso en embarazo y mujeres con potencial de embarazo. Riesgos asociados con ácido valproico: En humanos: De acuerdo con los reportes publicados y sin publicar, el ácido valproico puede producir efectos teratogénicos, como defectos en el tubo neural (por ejemplo, espina bífida) en hijos de mujeres que reciben ácido valproico durante el embarazo. Hay datos que sugieren un aumento en la incidencia de malformaciones congénitas asociado con el uso de ácido valproico durante el embarazo cuando se comparó con otros fármacos antiepilépticos. Por consiguiente, el ácido valproico debe considerarse para mujeres en edad fértil sólo después de haber discutido exhaustivamente los riesgos con el paciente y de haberlos valorado contra los beneficios potenciales del tratamiento, así mismo haber dado a la mujer consejería sobre métodos anticonceptivos. Hay múltiples reportes en las publicaciones clínicas que indican que el uso de fármacos antiepilépticos durante el embarazo aumenta la incidencia de defectos de nacimiento. Por lo tanto, los fármacos antiepilépticos sólo deben administrarse a mujeres fértiles si han demostrado ser esenciales en el manejo de su enfermedad. Los datos que se describen a continuación se obtuvieron casi exclusivamente de mujeres que recibieron valproato para tratar la epilepsia. La incidencia de defectos del tubo neural en el feto puede aumentar en madres que reciben valproato durante el primer trimestre del embarazo. El centro para el control de enfermedades (CDC, por sus siglas en inglés) de Estados Unidos estimó que las mujeres expuestas al ácido valproico tienen un riesgo de 1 a 2% de tener hijos con espina bífida. Se han reportado otras anomalías congénitas (por ejemplo, defectos craneofaciales, malformaciones cardiovasculares, hipospadias y anomalías que abarcan diferentes sistemas corporales) compatibles e incompatibles con la vida. Datos de un estudio de meta-análisis (incluyendo registros y estudios de cohorte) han mostrado una mayor incidencia de malformaciones congénitas en niños nacidos de mujeres epilépticas expuestas a monoterapia con valproato durante el embarazo. La información disponible indica que este efecto es dosis-dependiente. El valproato puede reducir las calificaciones del IQ después de la exposición in utero. Estudios epidemiológicos han indicado que los niños expuestos a valproato in utero tienen calificaciones menores en pruebas cognitivas que los niños expuestos in utero a otro antiepiléptico u otro fármaco no antiepiléptico. El más grande de estos estudios es un estudio prospectivo llevado a cabo en Estados Unidos y el Reino Unido que encontró que la exposición fetal a valproato tiene una asociación dosis-dependiente con habilidades cognitivas reducidas en un rango de dominios a los 6 años de edad, comparado con otros fármacos antiepilépticos en monoterapia. Existen reportes de retraso del desarrollo, autismo y/o trastorno del espectro de autismo en hijos de mujeres expuestas al ácido valpróico durante el embarazo. La exposición in utero a productos de valproato se ha asociado con atrofia cerebral con grados variables de compromiso neurológico, incluyendo retraso en el desarrollo y daño psicomotor (ver Precauciones generales). Riesgos en el neonato: Mujeres embarazadas que toman valproato pueden desarrollar alteraciones de la coagulación, incluyendo trombocitopenia, hipofibrinogenemia, y/o disminución en otros factores de coagulación, que pueden resultar en complicaciones hemorrágicas en el neonato, incluyendo la muerte (ver Precauciones generales-Trombocitopenia). Si se usa valproato durante el embarazo, se deben monitorear cuidadosamente los parámetros de coagulación. Se reportó insuficiencia hepática que causó la muerte de un recién nacido y de un lactante después del uso de valproato durante el embarazo. Se han recibido reportes de hipoglucemia en neonatos cuyas madres tomaron valproato durante el embarazo. Se han reportado casos de hipotiroidismo en neonatos cuyas madres han tomado valproato durante el embarazo. Puede presentarse síndrome de abstinencia (como en particular, agitación, irritabilidad, hiperexcitabilidad, temblores, hipercinesia, trastornos de tonicidad, tremor, convulsiones y trastornos en la alimentación) en neonatos cuyas madres tomaron valproato durante el último trimestre del embarazo. Las pruebas para detectar defectos del tubo neural y otros defectos usando los procedimientos actuales deben considerarse una parte de la rutina prenatal y en mujeres en edad fértil que reciben valproato. Los fármacos antiepilépticos no deben suspenderse abruptamente en pacientes a quienes se administra el fármaco para prevenir crisis convulsivas mayores debido a la fuerte posibilidad de precipitar estado epiléptico con hipoxia asociada y amenaza para la vida. En casos individuales en los que la severidad y frecuencia del trastorno de crisis convulsivas son tales que suspender la dosis del medicamento no representa una amenaza seria para el paciente, se puede considerar suspender el fármaco antes del embarazo y durante el mismo, aunque no es posible afirmar con seguridad que incluso las crisis menores no representan cierto peligro para el embrión o feto en desarrollo. En animales: Los estudios en animales han demostrado teratogenia inducida por valproato. En ratones, ratas, conejos y monos se ha observado aumento en la frecuencia de malformaciones, retraso del crecimiento intrauterino y muerte después de la exposición prenatal al valproato. Las malformaciones del sistema esquelético son las anomalías estructurales más comunes producidas en animales de experimentación, pero se han observado defectos de cierre del tubo neural en ratones expuestos a concentraciones plasmáticas maternas a valproato que exceden 230 mg/mL (2.3 veces el límite superior normal del rango terapéutico en humanos para epilepsia) durante los periodos sensibles del desarrollo embrionario. La administración de una dosis oral de 200 mg/kg/día o mayor (50% de la dosis diaria máxima en humanos o mayor en mg/m2) a ratas preñadas durante la organogénesis produjo malformaciones (esqueléticas, cardiacas y urogenitales) y retraso del crecimiento en las crías. Estas dosis produjeron niveles plasmáticos máximos maternos de valproato de aproximadamente 340 mg/mL o mayores (3.4 veces o más el límite superior del rango terapéutico en humanos). Se han reportado deficiencias conductuales en crías de ratas a las que se administró una dosis de 200 mg/kg/día durante la mayor parte del embarazo. Una dosis oral de 350 mg/kg/día (aproximadamente dos veces la dosis diaria máxima en humanos en mg/m2) produjo malformaciones esqueléticas y viscerales en conejos expuestos durante la organogénesis. Se observaron malformaciones esqueléticas, retraso del crecimiento y muerte en monos rhesus después de la administración de una dosis oral de 200 mg/kg/día (igual a la dosis diaria máxima en humanos en mg/m2) durante la organogénesis. Esta dosis produjo niveles plasmáticos máximos maternos de valproato de aproximadamente 280 mg/mL (2.8 veces el límite superior del rango terapéutico en humanos). Lactancia: El valproato se excreta en la leche materna. Se ha reportado que las concentraciones en la leche materna son 1 a 10% de las concentraciones séricas. Se desconoce el efecto que esto tendría en un lactante alimentado con leche materna. Se debe considerar suspender la lactancia materna cuando se administra valproato semisódico a mujeres en etapa de lactancia (ver Precauciones generales).
Reacciones secundarias y adversas: Epilepsia: Crisis convulsivas parciales complejas (CPC): Con base en un estudio controlado con placebo de tratamiento complementario para crisis parciales complejas, valproato semisódico generalmente fue bien tolerado con la mayoría de los eventos adversos clasificados como de severidad leve a moderada. La intolerancia fue el motivo principal de suspensión en los pacientes tratados con valproato semisódico (6%), en comparación con 1% de los pacientes tratados con placebo. La tabla 1 enumera los eventos adversos derivados del tratamiento reportados por ≥ 5% de los pacientes tratados con valproato semisódico y para los cuales la incidencia fue mayor que en el grupo placebo, en el estudio controlado con placebo de tratamiento complementario de crisis convulsivas parciales complejas. Debido a que los pacientes también fueron tratados con otros fármacos antiepilépticos, en la mayoría de los casos no es posible determinar si los siguientes eventos adversos pueden atribuirse al valproato semisódico solo o a la combinación de valproato semisódico y otros fármacos antiepilépticos.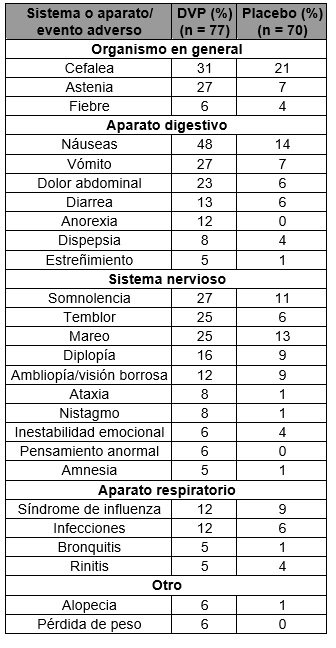
La tabla 2 enumera los eventos adversos derivados del tratamiento reportados por ≥ 5% de los pacientes en el grupo de dosis alta de valproato semisódico y para los cuales la incidencia fue mayor que en el grupo de dosis baja, en un estudio controlado de monoterapia con valproato semisódico para crisis parciales complejas. Debido a que durante la primera parte del estudio se estaba ajustando otro fármaco antiepiléptico para suspenderlo, en muchos casos no es posible determinar si los siguientes eventos adversos pueden atribuirse a valproato semisódico solo o a la combinación de valproato semisódico y otros fármacos antiepilépticos.
1 Cefalea fue el único evento adverso que se presentó en ≥ 5% de los pacientes en el grupo de dosis alta y con una incidencia igual o mayor en el grupo de dosis baja. Los siguientes eventos adversos fueron reportados por más de 1% pero menos de 5% de los 358 pacientes tratados con valproato semisódico en los estudios controlados de crisis convulsivas parciales complejas: Organismo en general: Dorsalgia, dolor precordial, malestar. Aparato cardiovascular: Taquicardia, hipertensión, palpitación. Aparato digestivo: Aumento del apetito, flatulencia, hematemesis, eructo, pancreatitis, absceso periodontal. Sangre y sistema linfático: Petequias. Trastornos metabólicos y alimenticios: ALT elevada, AST elevada. Sistema musculoesquelético: Mialgia, espasmos, artralgia, calambres en piernas, miastenia. Sistema nervioso: Ansiedad confusión, marcha anormal, parestesia, hipertonía, descoordinación, sueños anormales, trastorno de la personalidad. Aparato respiratorio: Sinusitis, aumento de tos, neumonía, epistaxis. Piel y anexos: Erupción cutánea, prurito, piel seca. Sentidos especiales: Disgeusia, visión anormal, sordera, otitis media. Aparato urogenital: Incontinencia urinaria, vaginitis, dismenorrea, amenorrea, frecuencia urinaria. Otras poblaciones de pacientes: A continuación se enumeran por sistema y aparato del organismo los eventos adversos que se han reportado con todas las formas farmacéuticas de valproato de estudios de epilepsia, reportes espontáneos y otras fuentes. Gastrointestinales: Los efectos reportados más comúnmente en el inicio del tratamiento son náusea, vómito e indigestión. Estos efectos generalmente son transitorios y rara vez es necesario suspender el tratamiento. Se han reportado diarrea, cólicos y estreñimiento y alteraciones gingivales (principalmente hiperplasia gingival). También se ha reportado anorexia con cierta pérdida de peso y aumento de apetito con aumento de peso. La administración de valproato semisódico con cubierta entérica puede resultar en la reducción de efectos secundarios gastrointestinales en algunos pacientes. Efectos en el SNC: Se han observado efectos sedantes en pacientes que reciben valproato solo pero se presentan con más frecuencia en pacientes que reciben tratamiento combinado. La sedación por lo regular disminuye al reducir el otro medicamento antiepiléptico. Se reportaron temblor (puede relacionarse con la dosis), alucinaciones, ataxia, cefalea, nistagmo, diplopía, asterixis, "manchas delante de los ojos", disartria, mareo, confusión, hipoestesia, vértigo, falta de coordinación, deterioro de la memoria, trastorno cognitivo y parkinsonismo con el uso de valproato. Se han presentado casos contados de coma en pacientes que reciben valproato solo o en combinación con fenobarbital. En contados casos se ha desarrollado encefalopatía con o sin fiebre poco después de la introducción de monoterapia con valproato sin evidencia de disfunción hepática o niveles plasmáticos de valproato inadecuadamente altos. A pesar de que se ha definido una recuperación después de retirar el fármaco, han habido muertes entre los pacientes con encefalopatía hiperamonémica, en particular en pacientes con trastornos subyacentes del ciclo de la urea (ver Trastornos del ciclo de la urea y Precauciones generales-Hiperamonemia-Topiramato). Ha habido reportes poscomercialización de atrofia cerebral y del cerebelo reversible e irreversible asociada temporalmente con el uso de productos de valproato. En algunos casos, los pacientes tuvieron secuelas permanentes. También ha habido reportes de atrofia cerebral con varias formas de problemas neurológicos incluyendo retraso en el desarrollo y deterioro psicomotor en niños que estuvieron expuestos in utero a productos de valproato (ver Precauciones generales-Atrofia cerebral y Restricciones de uso durante el embarazo y la lactancia). Dermatológicos: Pérdida de cabello transitoria, trastornos del cabello (tales como textura anormal, cambios en la coloración, crecimiento anormal), erupción cutánea, fotosensibilidad, prurito generalizado, eritema multiforme y síndrome de Stevens-Johnson. Se han reportado contados casos de necrólisis epidérmica tóxica incluyendo un caso mortal en un lactante de seis meses de edad que tomaba valproato y otros medicamentos concomitantes. Se reportó otro caso más de necrólisis epidérmica tóxica que causó la muerte a un paciente de 35 años con SIDA que tomaba diversos medicamentos concomitantes y con antecedentes de múltiples reacciones cutáneas a fármacos. Se han reportado reacciones cutáneas serias con la administración concomitante de lamotrigina y valproato (ver Interacciones medicamentosas y de otro género-Lamotrigina). Psiquiátricos: Alteración emocional, depresión, psicosis, agresión, hiperactividad psicomotora, hostilidad, agitación, trastorno en la atención, conducta anormal, trastorno de aprendizaje y deterioro de la conducta. Musculoesqueléticos: Debilidad: Se han recibido reportes de disminución de masa ósea, derivando posiblemente en osteoporosis y osteopenia, durante el tratamiento a largo plazo con medicamentos anticonvulsivos, incluyendo valproato. Algunos estudios indican que el calcio y vitamina D complementarios pueden ser benéficos para los pacientes que están en tratamiento crónico con valproato. Hematológicos: Trombocitopenia e inhibición de la fase secundaria de la agregación plaquetaria pueden reflejarse en alteración del tiempo de sangrado, petequias, contusiones, formación de hematomas, epistaxis y hemorragia (ver Precauciones generales e Interacciones medicamentosas y de otro género-Warfarina). Linfocitosis relativa, macrocitosis, hipofibrinogenemia, leucopenia, eosinofilia, anemia incluyendo macrocítica con o sin deficiencia de folato, inhibición de médula ósea, pancitopenia, anemia aplásica, agranulocitosis y porfiria aguda intermitente. Hepáticos: Las elevaciones menores de las transaminasas (por ejemplo, AST y ALT) y LDH son poco frecuentes y al parecer se relacionan con la dosis. Ocasionalmente, los resultados de pruebas de laboratorio incluyen aumentos de bilirrubina y cambios anormales en otras pruebas de función hepática. Estos resultados pueden reflejar una hepatotoxicidad potencialmente seria (ver Precauciones generales-Hepatotoxicidad). Endocrinos: Menstruaciones irregulares, amenorrea secundaria, aumento de mamas, galactorrea e hinchazón de las glándulas parótidas. Hiperandrogenismo (hirsutismo, virilismo, acné, alopecia con patrón masculino, y/o incremento andrógeno). Pruebas de función tiroidea anormales incluyendo hipotiroidismo (ver Precauciones generales). Existen reportes espontáneos poco frecuentes de enfermedad ovárica poliquística. No se ha establecido una relación de causa y efecto. Pancreáticos: Pancreatitis aguda incluyendo muertes (ver Precauciones generales-Pancreatitis). Metabólicos: Hiperamonemia (ver Precauciones generales), hiponatremia y secreción inadecuada de la hormona antidiurética (ADH). Existen reportes poco comunes de síndrome de Fanconi principalmente en niños. Se han reportado disminuciones en las concentraciones de carnitina aunque la relevancia clínica no se conoce. Se ha presentado hiperglicinemia (concentración elevada de glicina en plasma) y se relacionó con la muerte de un paciente con hiperglicinemia no cetósica preexistente. Genitourinarios: Enuresis, insuficiencia renal, nefritis túbulointersticial e infección de vías urinarias. Sentidos especiales: Se ha reportado pérdida de la audición, ya sea reversible o irreversible; sin embargo, no se ha establecido una relación de causa y efecto. También se ha reportado dolor de oído. Neoplasias benignas, malignas y no especificadas (incluyendo quistes y pólipos): Síndrome mielodisplásico. Trastornos respiratorios, torácicos y mediastinales: Derrame pleural. Otros: Reacción alérgica, anafilaxia, edema de extremidades, lupus eritematoso, rabdomiólisis, deficiencia de biotina/deficiencia de biotinidasa, dolor óseo, aumento de tos, neumonía, otitis media, bradicardia, vasculitis cutánea, fiebre e hipotermia. Manía: Aunque el valproato semisódico en cápsulas Sprinkle no ha sido evaluado en eficacia y seguridad en el tratamiento de episodios maniacos asociados con el trastorno bipolar, los siguientes eventos adversos fueron reportados en 1% o más de los pacientes en dos estudios clínicos controlados con placebo. Organismo en general: Dolor en cuello, rigidez de nuca, escalofríos. Sistema cardiovascular: Hipotensión, hipotensión postural, vasodilatación. Sistema digestivo: Incontinencia fecal, gastroenteritis, glositis. Sistema músculoesquelético: Artrosis. Sistema nervioso: Agitación, reacción catatónica, hipocinesia, hiperreflexia, discinesia tardía, vértigo. Piel y anexos: Furunculosis, rash maculopapular, seborrea. Sentidos especiales: Conjuntivitis, ojos secos, dolor ocular. Sistema urogenital: Disuria. Migraña: Aunque el valproato semisódico en cápsulas Sprinkle no ha sido evaluado en eficacia y seguridad en el tratamiento de profilaxis de migraña, los siguientes eventos adversos fueron reportados en 1% o más de los pacientes en dos estudios clínicos con valproato semisódico tabletas, controlados con placebo. Organismo en general: Edema facial. Sistema digestivo: Boca seca, estomatitis. Sistema urogenital: Cistitis, metrorragia, hemorragia vaginal.
Precauciones y relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Carcinogénesis: Se administró ácido valproico por vía oral a ratas Sprague Dawley y ratones ICR (HA/ICR) en dosis de 80 y 170 mg/kg/día (aproximadamente 10 a 50% de la dosis diaria máxima en humanos en mg/m2) durante dos años. En ambas especies se observó una variedad de neoplasias. Los principales hallazgos fueron un aumento estadísticamente significativo en la incidencia de fibrosarcomas subcutáneos en ratas macho que recibían dosis altas de ácido valproico y una tendencia relacionada con la dosis estadísticamente significativa a adenomas pulmonares benignos en ratones que recibían ácido valproico. No se conoce la importancia clínica de estos hallazgos en los humanos. Mutagénesis: El valproato no fue mutágeno en un análisis bacteriano in vitro (prueba de Ames), no produjo efectos letales dominantes en ratones y no aumentó la frecuencia de aberraciones cromosómicas en un estudio citogenético in vivo en ratas. En un estudio de niños epilépticos que tomaban valproato, se reportó aumento en la frecuencia de intercambio entre cromátidas hermanas (SCE), pero esta asociación no se observó en otro estudio realizado en adultos. Hay cierta evidencia de que el aumento en las frecuencias de SCE se relaciona con la epilepsia. Se desconoce la significancia biológica de un aumento en la frecuencia de SCE. Alteraciones de la fertilidad: Estudios de toxicidad crónica en ratas y perros jóvenes y adultos demostraron una reducción en la espermatogenia y atrofia testicular a dosis orales de 400 mg/kg/día o más altas en ratas (aproximadamente equivalente o superior a la dosis diaria máxima humana en mg/m2) y 150 mg/kg/día o superior en perros (aproximadamente 1.4 veces la dosis diaria máxima en humanos o superior en mg/m2). Los estudios de fertilidad del segmento I realizados en ratas demostraron que las dosis orales de hasta 350 mg/kg/día (aproximadamente igual a la dosis diaria máxima en humanos en mg/m2) durante 60 días no tienen efectos en la fertilidad. Se desconoce el efecto del valproato en el desarrollo testicular y en la producción de esperma y la fertilidad en humanos.
Interacciones medicamentosas y de otro género: Efectos en la depuración del valproato debidos a fármacos coadministrados: Los fármacos que afectan el nivel de expresión de enzimas hepáticas, en particular aquellos que elevan los niveles de glucuronosiltransferasa (tales como ritonavir), pueden aumentar la depuración de valproato. Por ejemplo, fenitoína, carbamazepina y fenobarbital (o primidona) pueden aumentar al doble la depuración de valproato. Por consiguiente, los pacientes en monoterapia generalmente tendrán vidas medias más prolongadas y concentraciones más altas que los pacientes que reciben politerapia con fármacos antiepilépticos. Por el contrario, los fármacos que son inhibidores de las isoenzimas del citocromo P450, por ejemplo, los antidepresivos, pueden tener poco efecto en la depuración de valproato porque la oxidación microsomal mediada por el citocromo P450 es una vía metabólica secundaria relativamente menor en comparación con la glucoronidación y beta oxidación. Debido a estos cambios en la depuración de valproato, el monitoreo de las concentraciones de valproato y los fármacos concomitantes se debe aumentar siempre que se introduzcan o retiren fármacos inductores de enzimas. La siguiente lista de sustancias proporciona información sobre la potencial influencia de éstas en la farmacocinética de valproato. La lista no es exhaustiva ni lo podría ser, ya que continuamente se reportan nuevas interacciones. Medicamentos en los que se han observado interacciones importantes potenciales: Ácido acetilsalicílico: Un estudio que incluyó la administración concomitante de ácido acetilsalicílico en dosis antipiréticas (11 a 16 mg/kg) con valproato a pacientes pediátricos (n = 6) reveló una disminución en la unión a proteínas y una inhibición del metabolismo de valproato. La fracción libre de valproato aumentó cuatro veces en presencia de ácido acetilsalicílico en comparación con valproato solo. La vía de b-oxidación que consiste en 2-E-ácido valproico, 3-OH-ácido valproico, y 3-ceto ácido disminuyó de 25% de metabolitos totales excretados en valproato solo a 8.3% en presencia de ácido acetilsalicílico. Se debe tener precaución si se debe administrar valproato y ácido acetilsalicílico concomitantemente. Antibióticos carbapenémicos: Se reportó una reducción clínicamente significativa en la concentración sérica de ácido valproico en pacientes que recibieron antibióticos carbapenémicos (ertapenem, imipenem, meropenem) y puede producir pérdida del control de crisis convulsivas. No se comprende bien el mecanismo de esta interacción. Las concentraciones séricas de ácido valproico deben monitorearse frecuentemente después de iniciar el tratamiento con carbapenem. Si las concentraciones séricas de ácido valproico disminuyen significativamente o si el control de crisis convulsivas se deteriora, debe considerarse un tratamiento antibacteriano o anticonvulsivo alternativo (ver Precauciones generales). Felbamato: Un estudio que incluyó la administración concomitante de 1,200 mg/día de felbamato con valproato a pacientes con epilepsia (n = 10) reveló un aumento en las concentraciones máximas plasmáticas promedio en 35% (de 86 a 115 mg/mL) en comparación con valproato solo. El incremento de la dosis de felbamato a 2,400 mg/día aumentó las concentraciones máximas plasmáticas promedio a 133 mg/mL (otro aumento de 16%). Al iniciar el tratamiento con felbamato puede ser necesario disminuir la dosis de valproato. Rifampicina: Un estudio que incluyó la administración de una dosis única de valproato (7 mg/kg) 36 horas después de cinco noches de dosificación diaria con rifampicina (600 mg) reveló un aumento de 40% en la depuración oral de valproato. Puede ser necesario ajustar la dosis de valproato cuando se administra concomitantemente con rifampicina. Fármacos en los que no se han observado interacciones ni probables interacciones clínicamente significativas: Antiácidos: Un estudio que incluyó la administración concomitante de valproato 500 mg con antiácidos comúnmente administrados (a base de sales de aluminio, entre otras) no reveló ningún efecto en el grado de absorción de valproato. Clorpromazina: Un estudio que incluyó la administración de 100 a 300 mg/día de clorpromazina a pacientes esquizofrénicos que ya recibían valproato (200 mg b.i.d.) reveló un aumento de 15% en los niveles mínimos plasmáticos de valproato. Haloperidol: Un estudio que incluyó la administración de 6 a 10 mg/día de haloperidol a pacientes esquizofrénicos que ya recibían valproato (200 mg b.i.d.) no reveló cambios significativos en los niveles mínimos plasmáticos de valproato. Cimetidina y ranitidina: Cimetidina y ranitidina no afectan la depuración de valproato. Efectos de valproato en otros fármacos: Se encontró que el valproato es un inhibidor débil de algunas isoenzimas P450, epóxido hidrasa y glucuroniltransferasas. La siguiente lista de sustancias proporciona información sobre el potencial de una influencia de la coadministración del valproato en la farmacocinética y farmacodinamia de diversos medicamentos comúnmente prescritos. La lista no es exhaustiva, ya que continuamente se reportan nuevas interacciones. Fármacos en los que se han observado interacciones potencialmente importantes con valproato: Amitriptilina/nortriptilina: La administración de una dosis oral única de 50 mg de amitriptilina a 15 voluntarios normales (diez varones y cinco mujeres) que recibieron valproato (500 mg b.i.d.) produjo una disminución de 21% en la depuración plasmática de amitriptilina y una disminución de 34% en la depuración neta de nortriptilina. Se han recibido contados reportes posteriores a la comercialización de que el uso concurrente de valproato y amitriptilina aumenta el nivel de amitriptilina. El uso concurrente de valproato y amitriptilina rara vez se ha asociado con toxicidad. Se debe considerar el monitoreo de los niveles de amitriptilina de pacientes que toman valproato concomitantemente con amitriptilina. Se debe considerar reducir la dosis de amitriptilina/nortriptilina en presencia del valproato. Carbamazepina/carbamazepina-10, 11-epóxido: Los niveles séricos de carbamazepina (CBZ) disminuyeron 17% mientras que el de carbamazepina-10, 11-epóxido (CBZ-E) aumentó en 45% al coadministrar valproato y CBZ a pacientes epilépticos. Clonazepam: El uso concomitante de ácido valproico y clonazepam puede inducir estado de ausencia en pacientes con antecedentes de crisis de ausencia. Diazepam: El valproato desplaza al diazepam de sus sitios de unión a la albúmina plasmática e inhibe su metabolismo. La administración concomitante de valproato (1,500 mg al día) aumentó la fracción libre de diazepam (10 mg) en 90% en voluntarios sanos (n = 6). La depuración plasmática y volumen de distribución de diazepam libre disminuyeron en 25 y 20%, respectivamente, en presencia de valproato. La vida media de eliminación de diazepam permaneció sin cambios al agregar valproato. Etosuximida: Valproato inhibe el metabolismo de etosuximida. La administración de una dosis única de etosuximida de 500