EVRA
JANSSEN-CILAG
Denominación genérica: Norelgestromina / Etinilestradiol.
Forma farmacéutica y formulación: Parche. Cada parche transdérmico contiene: Norelgestromina (NGMN) 6.00 mg. Etinilestradiol (EE) 0.60 mg. Excipiente cs parche.
Indicaciones terapéuticas: Anticoncepción femenina.
Farmacocinética y farmacodinamia: Evra es un parche transdérmico tipo matriz que consta de tres capas: La capa de soporte que está compuesta de una película flexible de color beige, constituida por una capa exterior de polietileno pigmentada de baja densidad y una capa interna de poliéster. Proporciona apoyo estructural y protege la capa adhesiva del medio ambiente. La capa intermedia contiene poli isobutileno/polibuteno adhesivo, crospovidona, poliéster no-tejido y lauril lactato como componentes inactivos. Los componentes activos en esta capa son las hormonas, norelgestromina y EE. La tercera capa protege la capa adhesiva durante el almacenamiento y se retira justo antes de la aplicación. Es un revestimiento transparente de tereftalato de polietileno (PET) con un recubrimiento de polidimetilsiloxano en el lado que está en contacto con la capa media adhesiva. Farmacocinética: Absorción: Después de la aplicación de EVRA®, tanto la norelgestromina como el etinilestradiol (EE) aparecen rápidamente en el suero, alcanzan una meseta en aproximadamente 48 horas, y se mantienen en un estado constante aproximado a lo largo del período de uso. Las concentraciones CSS para la norelgestromina y el etinilestradiol durante una semana de uso del parche son aproximadamente de 0.8 ng/ml y 50 pg/ml, respectivamente, y son generalmente consistentes en todos los estudios y sitios de aplicación. La absorción de norelgestromina y de etinilestradiol después de la aplicación de EVRA® en abdomen, glúteo, parte externa superior del brazo y torso superior (excluyendo senos) se evaluó en un estudio de diseño cruzado. Los resultados de este estudio indicaron que la CSS y el AUC para el glúteo, parte externa superior del brazo y torso superior para cada analito, fueron equivalentes. Los estrictos requerimientos de bioequivalencia para el AUC no se cumplieron en este estudio para el abdomen. Sin embargo, en un estudio farmacocinético por separado, paralelo, de aplicación múltiple, la CSS y el AUC para el glúteo y el abdomen no fueron estadísticamente diferentes. En un estudio de rango de dosis, EVRA® causó una supresión efectiva de la ovulación cuando se aplicó en el abdomen. Por lo tanto, los cuatro sitios son terapéuticamente equivalentes. La absorción de norelgestromina y de etinilestradiol después de la aplicación de EVRA® se estudió bajo las condiciones que se encuentran en un club deportivo (sauna, caminadoras y otros ejercicios aeróbicos) y en un baño de agua fría. Los resultados indicaron que para la norelgestromina no hubo efectos significativos del tratamiento sobre la CSS o el AUC cuando se comparó con el uso normal. Para el etinilestradiol, se observaron ligeros aumentos debido a las caminadoras y otros ejercicios aeróbicos. No hubo un efecto significativo del agua fría sobre estos parámetros. Los resultados de un estudio con EVRA® del uso prolongado de un solo parche anticonceptivo durante 7 días y 10 días indicó que las Css objetivo de norelgestromina y etinilestradiol se mantuvieron durante un período de 3 días de uso prolongado de EVRA® (10 días). Estos hallazgos sugieren que la eficacia clínica se mantiene incluso si se olvida un cambio programado hasta durante 2 días completos. Distribución: La norelgestromina y el norgestrel (un metabolito sérico de la norelgestromina) se unen en gran medida ( > 97%) a las proteínas séricas. La norelgestromina se une a la albúmina y no a la SHBG, mientras que el norgestrel se une principalmente a la SHBG, lo que limita su actividad biológica. El etinilestradiol se une extensamente a la albúmina sérica. Biotransformación: Como el parche EVRA® se aplica transdérmicamente, se evita el metabolismo de primer paso (vía el tracto gastrointestinal y/o el hígado) de la norelgestromina y del etinilestradiol que se espera después de la administración oral. Se da el metabolismo hepático de norelgestromina y sus metabolitos incluyen el norgestrel, el cual se une en gran medida a la SHBG, y varios metabolitos hidroxilados y conjugados. El etinilestradiol también se metaboliza en varios productos hidroxilados y a sus conjugados glucurónido y sulfato. Eliminación: Después del retiro de los parches, la cinética de eliminación de norelgestromina y de etinilestradiol fueron consistentes para todos los estudios con valores de la vida media de aproximadamente 28 horas y 17 horas, respectivamente. Los metabolitos del norelgestromina y de EE son eliminados por la vía renal y fecal. Linearidad/No-Linearidad: En estudios de dosis múltiples, se encontró que la CSS y el AUC se incrementan ligeramente con el tiempo cuando se comparan con la Semana 1 del Ciclo 1. En un estudio de tres ciclos, estos parámetros farmacocinéticos alcanzaron las condiciones del estado constante durante las tres semanas del Ciclo 3. Estas observaciones son indicativas de la cinética lineal denorelgestromina y de EE en el uso de EVRA®. Anticonceptivos transdérmicos contra orales: Los perfiles farmacocinéticos de los anticonceptivos hormonales combinados transdérmicos y orales son diferentes, y debe tenerse precaución cuando se haga una comparación directa de estos parámetros FC. En un estudio que comparó EVRA® con un anticonceptivo oral conteniendo NGM 250 g/EE 35 g, los valores de la Cmáx fueron 2 veces más elevados para NGMN y EE en sujetos a los que se administró el anticonceptivo oral en comparación con el EVRA®, mientras que la exposición global (AUC y Css) fue comparable en los sujetos tratados con EVRA®. La variabilidad inter-sujetos (CV %) para los parámetros FC después de la liberación de EVRA® fue mayor con relación a la variabilidad determinada con el anticonceptivo oral. Datos Pre-clínicos de Seguridad. Los datos preclínicos no revelan ningún riesgo especial para humanos, con base en estudios convencionales de seguridad, farmacología, toxicidad de dosis repetida, genotoxicidad, potencial carcinogénico, toxicidad para la reproducción. Los estudios realizados para examinar los efectos dérmicos de EVRA® indican que este sistema no tiene ningún potencial para producir sensibilización y sólo resulta en una irritación leve cuando se aplicó a piel de conejo. Farmacodinamia: EVRA® actúa a través del mecanismo de la supresión de gonadotropinas mediante las acciones estrogénicas y progestacionales de etinilestradiol y norelgestromina. El mecanismo primario de acción es la inhibición de la ovulación, aunque las alteraciones del moco cervical, la movilidad de las trompas de falopio y del endometrio también pueden contribuir a la eficacia del producto. Los estudios de la globulina de unión a hormonas sexuales (SHBG) y receptores, así como los estudios en animales y humanos, han demostrado que tanto el norgestimato (NGM) como la norelgestromina, el principal metabolito sérico del norgestimato, después de la administración oral, muestran una elevada actividad progestacional con una androgenicidad intrínseca mínima, lo cual ilustra la acción selectiva de EVRA®. La norelgestromina administrada de forma transdérmica en combinación con el etinilestradiol no contrarresta los incrementos inducidos por estrógenos en la SHBG, dando como resultado niveles más bajos de testosterona libre en suero en comparación con la línea base. Los siguientes beneficios, no anticonceptivos, para la salud, relacionados con el uso de anticonceptivos hormonales combinados, están soportados por estudios epidemiológicos que utilizaron en gran medida formulaciones contraceptivas hormonales que contienen estrógeno en dosis mayores a 35 microgramos de EE o 50 microgramos de mestranol. Efectos sobre la menstruación: Mayor regularidad del ciclo menstrual: Menor pérdida de sangre y menor incidencia de anemia por deficiencia de hierro Menor incidencia de dismenorrea. Efectos relacionados con la inhibición de la ovulación: Menor incidencia de quistes de ovario funcionales. Menor incidencia de embarazos ectópicos. Otros efectos: Menor incidencia de fibroadenomas y enfermedad fibroquística mamaria. Menor incidencia de enfermedad pélvica inflamatoria aguda. Menor incidencia de cáncer del endometrio. Menor incidencia de cáncer de ovarios
Contraindicaciones: EVRA® no debe usarse en mujeres que actualmente presenten las siguientes condiciones: Tromboflebitis, trastornos tromboembólicos. Antecedentes de tromboflebitis de venas profundas o trastornos tromboembólicos. Condiciones trombofilicas conocidas. Enfermedad cerebro-vascular o de arterias coronarias. Enfermedad cardiaca valvular con complicaciones103. Hipertensión severa (valores persistentes de 160+/100+ mmHg). Diabetes con implicación vascular. Migraña con aura focal. Carcinoma de mama conocido o sospecha del mismo. Carcinoma del endometrio u otra neoplasia conocida o sospechosa dependiente de estrógenos. Sangrado transvaginal anormal no diagnosticado. Antecedente familiar de ictericia colestática, ó ictericia idiopática crónica. Enfermedad hepatocelular aguda o crónica con función hepática anormal. Adenomas o carcinomas hepáticos. Embarazo conocido o sospecha del mismo. Hipersensibilidad a cualquiera de los componentes de este producto
Precauciones generales: Tabaquismo y Edad: El tabaquismo aumenta el riesgo de eventos cardiovasculares serios con el uso de anticonceptivos orales. Este riesgo aumenta con la edad (especialmente en mujeres de más de 35 años de edad) y con el número de cigarrillos fumados. Por esta razón, los anticonceptivos hormonales, incluyendo EVRA® no deben ser usados por mujeres de más de 35 años de edad que fuman. Peso Corporal ≥90 kg: El análisis de los datos de la fase III sugiere que EVRA® puede ser menos efectivo en usuarias con un peso corporal ≥90 kg que en usuarias con pesos corporales menores. Por debajo de 90 Kg, no hubo asociación entre el peso corporal y el embarazo. General: En caso de sangrado vaginal anormal persistente o recurrente, no diagnosticado, se deben tomar las medidas apropiadas para descartar la posibilidad de tumores malignos. Cuando se utilizó EVRA® correctamente en los estudios clínicos, la posibilidad de embarazo fue menor del 1% en el primer año de uso. La posibilidad de embarazo aumenta con los errores en la administración. Condiciones Pre-existentes: Cuando se ponderan los riesgos/beneficios del uso de anticonceptivos hormonales, el médico debe conocer las siguientes condiciones que pueden aumentar el riesgo de complicaciones asociadas con el uso de anticonceptivos hormonales: Condiciones que pueden aumentar el riesgo de desarrollo de complicaciones tromboembólicas venosas, p. ej., la inmovilización prolongada o cirugía mayor o cirugía de miembros pélvicos, obesidad o antecedentes familiares de enfermedad tromboembólica. Factores de riesgo para la enfermedad arterial, p. ej., tabaquismo, hiperlipidemia, hipertensión u obesidad. Migraña severa sin aura. Diabetes mellitus. Depresión severa o antecedentes de esta condición, Presencia o historia de colelitiasis. Ictericia idiopática crónica. Antecedentes familiares de ictericia colestática (p. ej., Rotor, Síndrome de Dubin-Johnson). Enfermedad Tromboembólica y otros Trastornos Vasculares: Un riesgo aumentado de tromboembolia y enfermedad trombótica que podría llevar a una discapacidad permanente o a la muerte se ha asociado con el uso de anticonceptivos hormonales y está bien establecida. Los estudios de control de casos han encontrado que el riesgo relativo de las usuarias en comparación con las no usuarias es de 3 para el primer episodio de trombosis venosa superficial, de 4 a 11 para trombosis de venas profundas o embolismo pulmonar, y de 1.5 a 6 para usuarias con factores predisponentes para enfermedad tromboembólica venosa. Los estudios han demostrado que el riesgo relativo es un poco más bajo, alrededor de 3 para los nuevos casos y alrededor de 4.5 para los nuevos casos que requieren hospitalización. El riesgo de enfermedad tromboembólica asociada con anticonceptivos hormonales no está relacionada con la duración del uso y desaparece después de que se suspende el uso del anticonceptivo hormonal. Se realizaron estudios epidemiológicos de casos-control en Estados Unidos, usando datos de quejas al sector salud para evaluar el riesgo de tromboembolismo venoso (TEV) en mujeres de 15-44 años que utilizaron ORTHO EVRA (Un parche transdérmico bioequivalente a EVRA) comparado con mujeres que utilizaron anticonceptivos orales que contenían 30-35 mcg de etinilestradiol (EE) y norgestimato (NGM) o levonorgestrel (LNG). El norgestimato es el profarmaco de la norelgestromina, la progestina de ORTHO EVRA®. Estos estudios (ver tabla 1) utilizaron diseños ligeramente diferentes y reportaron una razón de momios en rangos de 0.9 (indicando ningún incremento en el riesgo) a 2.5 (indicando un incremento en el riesgo de casi el doble). Algunos de los datos de los estudios epidemiológicos sugieren un aumento del riesgo de TEV con el uso de ORTHO EVRA® comparación con el uso de algunos anticonceptivos orales combinados. En un estudio se incluyó la revisión del diagrama de la paciente para confirmar la ocurrencia de TEV. Se realizaron dos estudios por parte del Programa de vigilancia medicamentosa de la colaboración de Boston (BCDSP) utilizando diferentes bases de datos con anticonceptivos orales que contenían levonorgestrel como comprador.
Como con otros anticonceptivos hormonales, el médico debe advertir a su paciente sobre cualquier manifestación temprana de alteraciones tromboembolicas (tromboflebitis, TEC, incluyendo embolismo pulmonar, alteraciones cerebrovasculares, trombosis en la retina). Si se presenta alguno de estos, Evra® debe descontinuarse inmediatamente. Se ha reportado el aumento de dos a cuatro veces en el riesgo relativo de complicaciones tromboembólicas post-operatorias con el uso de anticonceptivos hormonales. El riesgo relativo de trombosis venosa en las usuarias que tienen condiciones predisponentes es de dos veces el de las usuarias sin dichas condiciones médicas. Si es posible, deben suspenderse los anticonceptivos hormonales al menos cuatro semanas antes de y durante dos semanas después de una cirugía planeada del tipo asociado con un aumento en el riesgo de tromboembolismo y durante y después de una inmovilización prolongada, como el período inmediato post-parto o post-aborto también está asociado con un aumento en el riesgo de tromboembolia, los anticonceptivos hormonales deben iniciarse como se describe en las secciones "Uso después del parto" y "Uso después de un aborto o un aborto espontáneo". El riesgo relativo de trombosis arterial (p. ej., Ataque, infarto al miocardio) se incrementa por la presencia de otros factores predisponentes como el tabaquismo, la hipertensión, la hipercolesterolemia, la obesidad, la diabetes, antecedentes de toxemia pre-eclámptica y la edad. Los anticonceptivos hormonales han estado asociados con estas complicaciones vasculares serias. El riesgo de enfermedad vascular puede ser menos severo con las formulaciones de anticonceptivos hormonales que contienen dosis más bajas de estrógenos y progestágenos, aunque esto no se ha establecido de manera concluyente. El riesgo de efectos colaterales cardiovasculares serios aumenta con la edad y con un tabaquismo fuerte y es muy marcado en fumadoras mayores de 35 años de edad. A las usuarias de anticonceptivos hormonales se les debe recomendar enérgicamente que no fumen. Debido a la vaga sintomatología de muchos eventos tromboembólicos venosos, debe considerarse la suspensión de anticonceptivos hormonales en casos de sospecha de trombosis mientras se persiguen las intervenciones de diagnóstico. Ha habido reportes clínicos de trombosis de la retina asociada con el uso de anticonceptivos hormonales. Los anticonceptivos hormonales deben suspenderse si hay una pérdida de visión parcial o completa inexplicable: aparición de proptosis o diplopía; papiledema o lesiones vasculares de la retina. Deben tomarse inmediatamente medidas diagnósticas y terapéuticas apropiadas. Hipertensión: Se ha reportado el incremento en la presión sanguínea en algunas usuarias que toman anticonceptivos hormonales. Los estudios indican que este incremento es más probable que se presente en usuarias de anticonceptivos hormonales mayores y con una duración en el uso extendida. Para muchas usuarias, la presión sanguínea elevada volverá a lo normal después que dejen de tomar los anticonceptivos hormonales. No hay diferencia en la presencia de la hipertensión entre las usuarias antiguas y las que nunca han usado anticonceptivos. En tres estudios de anticoncepción de EVRA® (n = 1530, 819, y 748, respectivamente) la media de los cambios a partir de la línea base en la presión sanguínea sistólica y diastólica fue de menos de 1 mmHg. Las usuarias con hipertensión deben tener su condición bajo control antes de poder iniciar una terapia con anticonceptivos hormonales. La terapia con anticonceptivos hormonales debe suspenderse si ocurre una elevación significativa de la presión sanguínea (≥160/100). Enfermedad hepatobiliar: Los adenomas hepáticos benignos están asociados con el uso de anticonceptivos hormonales combinados. Los cálculos indirectos han estimado que el riesgo atribuible está en el rango de 3.3 casos/100,000 para las usuarias, un riesgo que aumenta después de 4 o más años de uso, especialmente con los anticonceptivos hormonales que contienen 50 microgramos o más de estrógenos. La ruptura de los adenomas hepáticos benignos puede causar la muerte debido a la hemorragia intra-abdominal. Los estudios han demostrado que las usuarias de anticonceptivos hormonales combinados tienen un riesgo aumentado de desarrollar carcinoma hepatocelular. Las alteraciones de la vesicular biliar incluyen colecistitis y colelitiasis, han sido reportadas con el uso de anticonceptivos hormonales. Carcinoma de los Órganos Reproductores y las Mamas: La mayoría de los estudios sugieren que el uso de anticonceptivos hormonales no está asociado con un aumento global en el riesgo de desarrollar cáncer de mama. Algunos estudios han reportado un aumento del riesgo relativo de desarrollar cáncer de mama, particularmente en una edad más joven. Se ha reportado que este riesgo relativo aumentado está relacionado con la duración del uso, antes del primer embarazo. Un meta-análisis de 54 estudios epidemiológicos reporta que las usuarias que están usando anticonceptivos hormonales combinados o los han usado en los últimos 10 años, están en un riesgo ligeramente mayor de tener cáncer de mama diagnosticado, aunque los cánceres adicionales tienden a estar confinados en la mama. No es posible inferir de estos datos si los patrones de riesgo observados se deben a un diagnóstico temprano de cáncer de mama en usuarias de tiempo atrás, a los efectos biológicos de los anticonceptivos hormonales o a una combinación de ambos factores. Este meta-análisis también sugiere que la edad a la cual las usuarias suspenden el uso de anticonceptivos hormonales combinados es un factor de riesgo importante para el cáncer de mama; a mayor edad se suspenden, es mayor el número de cánceres de mama diagnosticados. La duración del uso se consideró menos importante. El posible incremento en el riesgo de cáncer de mama debe discutirse con las usuarias y ponderarse contra los beneficios de los anticonceptivos hormonales combinados, tomando en cuenta la evidencia de que ofrecen una protección sustancial contra el riesgo de desarrollar cáncer de ovarios y del endometrio. Algunos estudios sugieren que el uso de anticonceptivos hormonales ha sido asociado con un mayor riesgo de neoplasia cervical intraepitelial en algunas poblaciones de usuarias. Sin embargo, continúa habiendo controversia acerca de hasta dónde pueden deberse dichos hallazgos a las diferencias en el comportamiento sexual y a otros factores. Efectos Metabólicos: Los anticonceptivos hormonales pueden causar una reducción en la tolerancia a la glucosa. Se ha demostrado que este efecto está directamente relacionado con la dosis de estrógenos. Los progestágenos aumentan la secreción de insulina y crean resistencia a la insulina. Este efecto varía con los diferentes agentes progestacionales. Sin embargo, en las mujeres no diabéticas, los anticonceptivos hormonales parecen no tener efecto sobre la glucosa sanguínea en ayunas. Debido a estos efectos demostrados, las usuarias pre-diabéticas y diabéticas en particular deben ser monitoreadas cuidadosamente mientras utilizan anticonceptivos hormonales. Una pequeña proporción de mujeres tendrá una hipertrigliceridemia persistente mientras toma anticonceptivos hormonales. Se han reportado cambios en los triglicéridos séricos y en los niveles de lipoproteínas en las usuarias de anticonceptivos hormonales. Cefalea: Al igual que con todos los anticonceptivos hormonales, los siguientes eventos requieren la suspensión de EVRA® y la evaluación de la causa: aparición o exacerbación de migrañas con o sin aura focal; o desarrollo de cefaleas con un nuevo patrón que es recurrente, persistente o severo. Sangrado Irregular: El sangrado intermenstrual, el manchado y/o la amenorrea pueden encontrarse en las usuarias de anticonceptivos hormonales, especialmente durante los primeros 3 meses de uso. Deben considerarse las causas no hormonales y, si es necesario, tomar medidas de diagnóstico adecuadas para descartar una enfermedad orgánica o el embarazo. Algunas usuarias pueden experimentar amenorrea u oligomenorrea después de suspender la anticoncepción hormonal, especialmente cuando dicha condición era pre-existente. Cloasma: El cloasma puede presentarse ocasionalmente con el uso de anticonceptivos hormonales, especialmente en usuarias con antecedentes de cloasma gravidarum. Las usuarias con tendencia al cloasma deben evitar la exposición al sol o a la radiación ultravioleta cuando están utilizando EVRA®. El cloasma con frecuencia no es completamente reversible. Anticonceptivos transdérmicos contra orales: Quienes prescriben deben estar informados de las diferencias en los perfiles farmacocinéticos (FC) de los anticonceptivos hormonales combinados transdérmicos y orales y deben tener precaución cuando hacen una comparación directa entre estos parámetros. En general, los parches transdérmicos están diseñados para mantener una liberación constante de EE y NGMN durante un período de siete días, mientras que los anticonceptivos orales se administran diariamente y producen máximos y mínimos diarios. La variabilidad inter-sujetos (CV %) para los parámetros FC después de la liberación del parche es mayor con relación a la variabilidad determinada en los anticonceptivos orales. La relevancia clínica de las diferencias en los perfiles FC entre la liberación transdérmica y oral no se conoce. (Ver sección Propiedades farmacocinéticas - Anticonceptivos transdérmicos contra orales.)
Restricciones de uso durante el embarazo y la lactancia: No se use en el embarazo y la lactancia. EVRA® está contraindicado durante el embarazo. Los estudios epidemiológicos indican que no hay un aumento del riesgo de defectos de nacimiento en niños nacidos de mujeres que utilizaron anticonceptivos hormonales antes del embarazo. La mayoría de los estudios recientes tampoco indican un efecto teratogénico, particularmente en lo que se refiere a anomalías cardíacas y a defectos de reducción de extremidades, cuando se usan anticonceptivos hormonales inadvertidamente durante el inicio del embarazo. Una pequeña cantidad de los esteroides anticonceptivos y/o de sus metabolitos puede excretarse a través de la leche materna. Se han identificado cantidades pequeñas de esteroides anticonceptivos hormonales combinados en la leche de madres lactantes y se han reportado unos cuantos efectos adversos en el niño, incluyendo ictericia y agrandamiento del pecho. Adicionalmente, los anticonceptivos hormonales combinados administrados en el período post-parto pueden interferir con la lactancia reduciendo la cantidad y la calidad de la leche materna. Si es posible, debe aconsejarse a la madre que amamanta que no utilice EVRA® u otro anticonceptivo hormonal combinado, sino que use otras formas de anticoncepción hasta que el niño haya sido destetado completamente.
Reacciones secundarias y adversas: Información de estudios clínicos: La seguridad de EVRA® fue evaluada en 3,330 mujeres sexualmente activas que participaron en tres estudios fase clínica III, los cuales fueron diseñados para evaluar la eficacia anticonceptiva. Las pacientes recibieron seis ó 13 ciclos de anticonceptivos (EVRA® o anticonceptivos orales como comparador), tomaron por lo menos una dosis del medicamento de estudio y proporcionaron la información respectiva de seguridad. Los eventos adversos reportados con mayor frecuencia durante los estudios clínicos fueron: síntomas mamarios, cefalea, alteraciones del sitio de aplicación y nausea. Los eventos adversos más comunes que ocasionaron descontinuación del estudio fueron: reacción del sitio de aplicación, síntomas mamarios (Incluyendo molestias mamarias, congestión mamaria y dolor mamario), nausea, cefalea y labilidad emocional. Las reacciones adversas (RAD) reportadas en 1% de los pacientes tratados con EVRA® en los estudios clínicos se muestran en la tabla 2.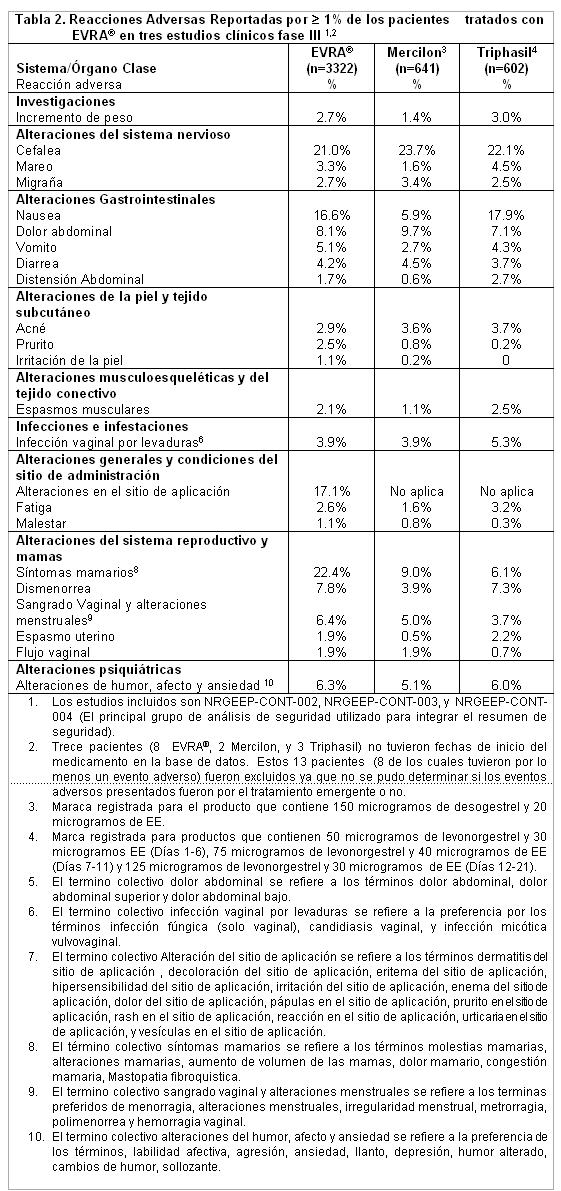
Ls reacciones adversas adicionales que ocurrieron en < 1% de los sujetos tratados con EVRA® en los estudios clínicos mencionados se listan en la Tabla 3.
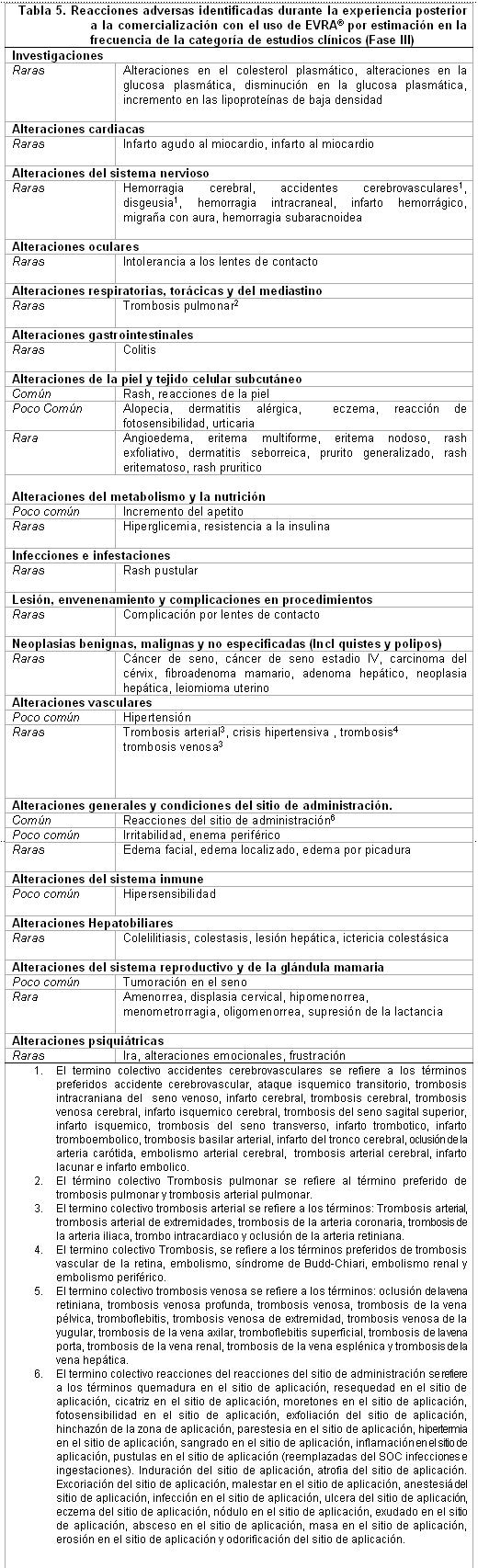
Datos Post-Comercialización: Las reacciones adversas adicionales identificadas en primera instancia posterior a la experiencia post-comercialización con el uso de EVRA®, se incluyen en la tabla 4 y tabla 5. En cada tabla, las frecuencias son proporcionadas de acuerdo a la siguiente convención. Muy común ≥1/10. Común ≥1/100 y < 10. Poco común ≥1/1,000 y < 1/100. Raras ≥1/10,000 y < 1/1,000. Muy raras < 1/10,000, incluyendo reportes aislados. En la tabla 4, las reacciones adversas están presentadas por frecuencia de la categoría sobre los reportes espontáneos, mientras que en la tabla 5, las mismas Reacciones Adversas están presentadas basadas en la frecuencia de la categoría sobre la incidencia en los estudios clínicos o epidemiológicos cuando se es conocido.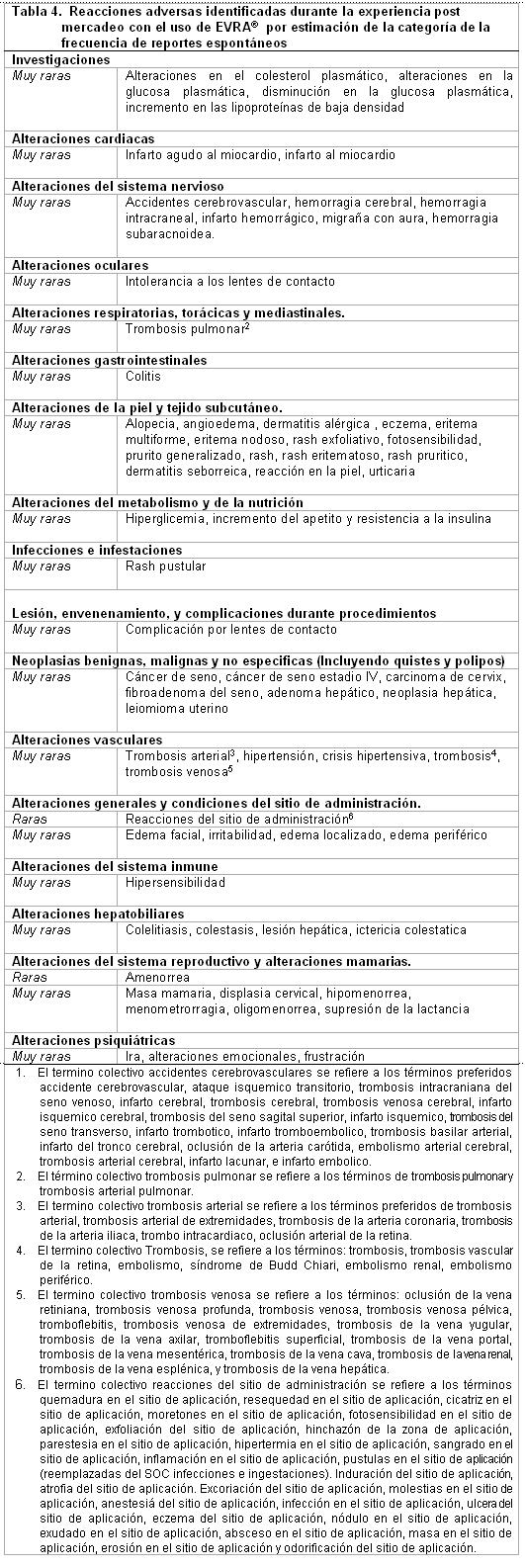
Interacciones medicamentosas y de otro género: Cambios en la eficacia anticonceptiva asociada con la administración concomitante de otros medicamentos: La administración de anticonceptivos hormonales con medicamentos o plantas, que inducen enzimas metabolizadas por CYP3A4, que metaboliza hormonas anticonceptivas, deben ser aconsejadas en la utilización de anticonceptivos adicionales u otra forma de anticoncepción. Los medicamentos o hierbas que inducen tales enzimas pueden producir disminución en las concentraciones plasmáticas de los anticonceptivos hormonales y pueden reducir la efectividad del anticonceptivo hormonal o incrementar el sangrado intermenstrual. Algunos medicamentos o hierbas que pueden disminuir la efectividad de los anticonceptivos hormonales incluyen: Algunos antiepilepticos (ej. Carbamazepina, acetato de eslicarbazepina, felbamato, oxcarbazepina, fenitoina, rufinamida, topiramato). (Fos)aprepitant. Barbitúricos. Bosentan. Griseofulvina. Algunos (o combinaciones de) inhibidores de proteasa del VIH (ej. Nelfinavir, ritonavir, inhibidores de proteasa potenciados con ritonavir). Modafinil. Algunos inhibidores de transcriptasa reversa no nucleosida (ej. nevirapina). Rifampicina y rifabutina. Hierba de San Juan. Incremento en los niveles de hormonas plasmáticas asociado con la administración concomitante de medicamentos: Algunos medicamentos y jugo de toronja pueden incrementar los niveles en plasma del etinilestradiol si son administrados concominantemente. Algunos ejemplos incluyen: Acetaminofen. Ácido ascórbico. Inhibidores del CYP3A4 (incluyendo itraconzaol, ketoconazol, voriconazol, fluconazol y jugo de toronja). Etoricoxib. Algunos inhibidores de protease de VIH (ej. atazanavir, indinavir). Inhibidores de la HMG-coA reductasa (incluyendo atorvastatina y rosuvastatina). Algunos inhibidores de transcriptasa reversa no nucleosida (ej. nevirapina). Cambios en los niveles plasmáticos de los medicamentos administrados concomitantemente: La combinación de anticonceptivos hormonales puede también afectar la farmacocinética de algunos medicamentos si se utilizan concominantemente. Ejemplos de medicamentos en los que se puede incrementar los niveles plasmáticos (debido a una inhibición del CYP) incluyen: Ciclosporina. Omeprazol. Prednisona. Selegilina. Teofillina. Tizanidina. Voriconazol. Ejemplos de medicamentos en los que se puede disminuir los niveles plasmáticos (por inducción de glucoronidación) incluyen: Acetaminofen. Ácido clofibrico. Lamotrigina. Morfina. Ácido salicílico. Temazepam. Lamotrigina: Los anticonceptivos hormonales combinados han demostrado disminuir significativamente las concentraciones plasmáticas de lamotrigina cuando son administrados concomitantemente probablemente por inducir la glucoronidación de lamotrigina. Esto podría reducir el control de las convulsiones, por consiguiente se requiere de un ajuste de la lamotrigina. Los médicos deben ser advertidos de consultar la información para prescribir con respecto al uso concomitante de medicamentos para obtener mayor información acerca de las interacciones de los anticonceptivos hormonales o del potencial de alteraciones enzimáticas y la posible necesidad de ajuste de dosis.
Alteraciones en los resultados de pruebas de laboratorio: Ciertas pruebas endocrinas y de funcionamiento hepático, así como diversos componentes sanguíneos, pueden ser afectados por los anticonceptivos hormonales: Aumento de la protrombina y de los factores VII, VIII, IX y X; disminución de la antiprotrombina III; reducción de proteína S; mayor agregación plaquetaria inducida por la norepinefrina (noradrenalina). Aumento de la globulina fijadora de tiroxina (TBG), que lleva a un incremento de la hormona tiroidea total circulante, medida por el yodo proteico (PBI), T4 por columna o por radioinmunoensayo. La captación de resina T3 libre decrece, reflejando el aumento de TBG; la concentración de T4 libre no se altera. Otras proteínas fijadoras pueden aumentar en el suero. Las globulinas fijadoras de hormonas sexuales (SHBG) aumentan, lo cual provoca mayores niveles de esteroides sexuales endógenos circulantes totales. Sin embargo, los niveles de esteroides sexuales libres o biológicamente activos se reducen o permanecen invariables. La lipoproteína de alta densidad (HDL-C), el colesterol total (C total), la lipoproteína de baja densidad (LDL-C) y los triglicéridos pueden aumentar ligeramente con EVRA®, mientras que la relación LDL-C/HDL-C puede permanecer invariable. La tolerancia a la glucosa puede disminuir. Los niveles de folato sérico pueden abatirse debido a la terapia anticonceptiva hormonal. Esto tiene el potencial de ser clínicamente significativo si una mujer se embaraza justo después de suspender los anticonceptivos hormonales. Ahora, a todas las mujeres se les aconseja tomar un complemento de ácido fólico antes y después de la concepción.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Los estudios epidemiológicos no indican mayor riesgo de defectos congénitos en hijos de mujeres que emplearon anticonceptivos hormonales antes del embarazo. La mayoría de los estudios recientes tampoco indican un efecto teratogénico, particularmente en cuanto a anomalías cardiacas y defectos de reducción de extremidades se refiere, cuando se emplean inadvertidamente anticonceptivos hormonales tempranamente en el embarazo. Neoplasias hepáticas: Los adenomas hepáticos benignos están asociados con el uso de anticonceptivos hormonales combinados. Cálculos indirectos han estimado que para las usuarias el riesgo atribuible está en el rango de 3.3 casos/100,000, un riesgo que aumenta después de 4 o más años de uso, especialmente con anticonceptivos hormonales que contienen 50 microgramos o más de estrógeno. La ruptura de adenomas hepáticos benignos puede causar la muerte por hemorragia intraabdominal. Los estudios han mostrado que las usuarias de anticonceptivos hormonales combinados tienen un mayor riesgo de desarrollar carcinoma hepatocelular. Carcinoma de órganos reproductores y mamas: La mayoría de los estudios sugieren que el uso de anticonceptivos hormonales no está asociado con un aumento general en el riesgo de desarrollar cáncer de mama. Algunos estudios han reportado mayor riesgo relativo de cáncer de mama, particularmente a una edad joven. Se ha reportado que este aumento del riesgo relativo está relacionado con la duración de uso, antes del embarazo de primer término. El posible incremento del riesgo de cáncer de mama debe discutirse con las usuarias y ponderarse con los beneficios de los anticonceptivos hormonales combinados, tomando en cuenta la evidencia de que ellos ofrecen protección sustancial contra el riesgo de desarrollar cáncer ovárico y endometrial. Algunos estudios sugieren que el uso de anticonceptivos hormonales se ha asociado con un mayor riesgo de padecer neoplasia intraepitelial cervical en algunas poblaciones de usuarias. Sin embargo, sigue habiendo controversia respecto al grado en que tales hallazgos pueden deberse a las diferencias en el comportamiento sexual y a otros factores.
Dosis y vía de administración: EVRA® debe aplicarse sobre la piel sana intacta, limpia, seca y sin pelo sobre el glúteo, abdomen, en la parte superior externa del brazo o en la parte superior del torso, en un lugar donde no haya roce continuo con la ropa ajustada. EVRA® no debe colocarse sobre los pechos o sobre la piel que esté enrojecida, irritada o con cortes. Cada parche consecutivo de EVRA® debe aplicarse en un lugar diferente de la piel para ayudar a evitar una potencial irritación, aunque puede dejarse dentro del mismo sitio anatómico. El parche debe oprimirse con firmeza hasta que los bordes se adhieran bien. Para evitar la interferencia con las propiedades adhesivas de EVRA®, no debe aplicarse maquillaje, cremas, lociones, polvos u otros productos tópicos al área de la piel donde está colocado el parche EVRA® o donde se vaya a colocar. Se recomienda que la usuaria revise visualmente su parche diariamente para asegurarse de que continúa la adhesión apropiada. Para lograr la máxima eficacia anticonceptiva, EVRA® debe usarse exactamente como se indica. Sólo debe utilizarse un parche a la vez. La anticoncepción con EVRA® empieza el primer día de la menstruación. El día que se aplica el primer parche (Día 1/Día de Inicio) determina los Días de Cambio posteriores. El Día de Cambio del parche será ese mismo día de cada semana (Días del ciclo 8, 15, 22 y Día 1 del siguiente ciclo). Se aplica un solo parche y se utiliza durante una semana completa (7 días). Cada parche usado se retira y se sustituye inmediatamente con uno nuevo el mismo día de la semana (Día de Cambio) el día 8 y el Día 15 del ciclo. Los cambios del parche pueden darse a cualquier hora del Día de Cambio programado. La cuarta semana empieza el Día 22 sin parche. El nuevo ciclo de anticoncepción empieza el siguiente día después de la semana sin parche; el siguiente parche EVRA® debe aplicarse incluso si no ha habido sangrado o si el sangrado continúa todavía. Bajo ninguna circunstancia debe haber un intervalo de más de 7 días sin parche entre los ciclos de administración. Si se dejan pasar más de 7 días sin parche, es posible que la usuaria no esté protegida contra el embarazo. En este caso, puede utilizarse un contraceptivo no hormonal de manera concurrente durante 7 días. Al igual que con los anticonceptivos orales combinados, aumenta el riesgo de ovulación con cada día que transcurre del período recomendado sin contraceptivo. Si han ocurrido relaciones sexualesl durante dicho intervalo prolongado sin parche, debe considerarse la posibilidad de fertilización. Si la terapia del Ciclo 1 empieza después del Día 1 del ciclo menstrual, debe usarse de manera concurrente un anticonceptivo no hormonal solamente durante los primeros 7 días consecutivos del primer ciclo de tratamiento. Si los bordes del parche EVRA® se levantan o se separan completamente y permanecen separados, ocurre una liberación insuficiente del fármaco. Si EVRA® permanece incluso parcialmente separado: Durante menos de un día (hasta 24 horas): debe volverse a aplicar en el mismo lugar o reemplazarse con un nuevo parche EVRA® inmediatamente. No es necesario ningún anticonceptivo adicional. El siguiente parche EVRA® debe aplicarse el "Día de Cambio" acostumbrado. Durante más de un día (24 horas o más) o si la usuaria no se da cuenta cuando el parche está levantado o se ha desprendido: La usuaria puede no estar protegida contra el embarazo. La usuaria debe detener el ciclo anticonceptivo presente e iniciar inmediatamente un nuevo ciclo mediante la aplicación de un nuevo parche EVRA®. Ahora hay un nuevo "Día 1" y un nuevo "Día de Cambio". Debe usarse de manera concurrente un anticonceptivo no hormonal solamente durante los primeros 7 días del nuevo ciclo. El parche no debe volverse a aplicar si ya no tiene adhesión, si se ha pegado con otra parte del mismo parche o a otras superficies, o si tiene otro material pegado a él o si se ha desprendido o caído antes. Si un parche no puede volverse a pegar, debe aplicarse un nuevo parche inmediatamente. No deben usarse adhesivos complementarios o envolturas para mantener el parche EVRA® en su sitio. Si los Días de Cambio del parche EVRA® siguiente están retrasados: Al inicio de cualquier ciclo del parche (Semana Uno/Día 1): La usuaria puede no estar protegida contra el embarazo. La usuaria debe aplicar el primer parche del nuevo ciclo tan pronto como lo recuerde. Ahora hay un nuevo "Día de Cambio" del parche y un nuevo "Día 1". Debe usarse de manera concurrente un anticonceptivo no hormonal durante los primeros 7 días del nuevo ciclo. Si se han ocurrido relaciones sexuales durante dicho intervalo prolongado sin parche, debe considerarse la posibilidad de fertilización. A la mitad del ciclo (Semana Dos/Día 8 ó Semana Tres/Día 15): Durante uno o dos días (hasta 48 horas): la usuaria debe aplicarse un nuevo parche EVRA® inmediatamente. El siguiente parche EVRA® debe aplicarse el "Día de Cambio" acostumbrado. No se requiere del uso de un anticonceptivo adicional. Durante más de dos días (48 horas o más): La usuaria puede no estar protegida contra el embarazo. La usuaria debe s