EXOTIB DUO
SIEGFRIED
Denominación genérica: Ezetimiba, Simvastatina
Forma farmacéutica y formulación: Cada tableta contiene: Ezetimiba 10 mg. Simvastatina 20 mg Excipientes cbp 1 tableta
Indicaciones terapéuticas: Hipercolesterolemia primaria: EXOTIB DUO® está indicado como tratamiento agregado a la dieta para disminuir las concentraciones elevadas de colesterol total, colesterol de las lipoproteínas de baja densidad (C-LDL), apolipoproteína B (Apo B), triglicéridos y colesterol de las lipoproteínas distintas de las de alta densidad (C-no-HDL) y para aumentar el colesterol de las lipoproteínas de alta densidad (C-HDL) en pacientes adultos y adolescentes (10 a 17 años de edad) con hipercolesterolemia primaria (familiar heterocigótica y no familiar) o con hiperlipidemia mixta.Se puede agregar fenofibrato al tratamiento con EXOTIB DUO® en pacientes adultos con hiperlipidemia mixta que requieran de una mayor reducción de triglicéridos (TG) y C-no-HDL, así como de un incremento en el CHDL.Hipercolesterolemia familiar homocigótica: EXOTIB DUO® está indicado para disminuir las concentraciones elevadas de colesterol total y de C-LDL en los pacientes adultos y adolescentes (10 a 17 años de edad) con hipercolesterolemia familiar homocigótica, los cuales pueden recibir también tratamientos adjuntos (por ejemplo, LDL-aféresis).
Farmacocinética y farmacodinamia: Absorción: Ezetimiba: Tras su administración por vía oral, la ezetimiba es absorbida rápidamente y transformada extensamente por conjugación en un glucurónido fenólico con actividad farmacológica (glucurónido de ezetimiba). El glucurónido de ezetimiba alcanza el promedio de concentración plasmática máxima (Cmáx.) en una a dos horas, y la ezetimiba en cuatro a doce horas. No se puede determinar la biodisponibilidad absoluta de la ezetimiba, por ser ésta prácticamente insoluble en medios acuosos apropiados para ser inyectados. La administración concomitante de alimentos (altos en grasas o sin grasas) no tuvo ningún efecto sobre la biodisponibilidad de la ezetimiba administrada por vía oral en forma de tabletas con 10 mg. Simvastatina: Se halló que la disponibilidad del beta-hidroxiácido en la circulación sistémica después de una dosis oral de simvastatina es de menos de 5% de la dosis, lo cual concuerda a con una extensa extracción hepática de primer paso. Los principales metabolitos de la simvastatina presentes en el plasma humano son el beta-hidroxiácido y otros cuatro metabolitos activos.En comparación con la administración en ayunas, no se modificaron las concentraciones plasmáticas de los inhibidores tanto activos como totales cuando se administró la simvastatina inmediatamente después de una comida de prueba. Distribución: Ezetimiba: Se unen a las proteínas plasmáticas humanas 99.7% de la ezetimiba y 88 a 92% del glucurónido de ezetimiba. Semvastatina: Tanto la simvastatina como el beta-hidroxiácido se unen a las proteínas plasmáticas (95%).La farmacocinética de dosis únicas y múltiples de simvastatina mostró que no ocurrió ninguna acumulación del medicamento después de la administración de dosis múltiples. En todos esos estudios farmacocinéticos, la concentración máxima de los inhibidores en el plasma se alcanzó 1.3 a 2.4 horas espués de la dosis. Metabolismo: Ezetimiba: La ezetimiba es metabolizada principalmente en el intestino delgado y en el hígado por conjugación con el ácido glucurónico (una reacción de fase II), y después es excretada con la bilis. En todas las especies estudiadas se ha observado un metabolismo oxidativo mínimo (una reacción de fase I). La ezetimiba y el glucurónido de ezetimiba son las principales formas del medicamento que se detectan en el plasma; constituyen 10 a 20% y 80 a 90%, respectivamente, del medicamento total en el plasma. Ambas formas son eliminadas lentamente del plasma, con evidencia de un considerable reciclamiento enterohepático. La vida media de la ezetimiba y de su glucurónido es de 22 horas aproximadamente. Simvastatina: La simvastatina es una lactona inactiva que in vivo es transformada rápidamente por hidrólisis en el beta-hidroxiácido correspondiente, el cual es un potente inhibidor de la reductasa de la HMG-CoA. La hidrólisis ocurre principalmente en el hígado; la hidrólisis en el plasma humano es muy lenta. En el humano la simvastatina se absorbe bien y es sometida a una extracción hepática extensa de primer paso.La extracción en el hígado es dependiente del flujo sanguíneo hepático. El hígado es el sitio primario de acción, con excreción subsecuente de equivalentes del fármaco en la bilis. Consecuentemente, la disponibilidad del fármaco activo en la circulación sanguínea es bajo. Tras la inyección intravenosa del metabolito beta-hidroxiácido, su vida media fue de 1.9 horas en promedio. Eliminación: Ezetimiba: Tras la administración oral de 20 mg de 14C-ezetimiba a personas, la ezetimiba total representó aproximadamente 93% de la radiactividad total en el plasma.En un periodo de diez días se recuperó aproximadamente 78% de la radiactividad en las heces y 11% en la orina. A las 48 horas, no hubo radiactividad detectable en el plasma. Simvastatina: Después de administrar una dosis oral de simvastatina radiactiva a personas, éstas excretaron 13% de la radiactividad con la orina y 60% con las heces en 96 horas.La cantidad recuperada de las heces representa los equivalentes del medicamento absorbido y excretado en la bilis y el medicamento no absorbido. Tras una inyección intravenosa del metabolito beta-hidroxiácido, se excretó con la orina en forma de inhibidores sólo un promedio de 0.3% de la dosis administrada.Hubo un beneficio general para todos los accidentes cerebrovasculares, sin embargo, hubo un pequeño aumento no significativo en el accidente cerebrovascular hemorrágico en el grupo de ezetimibe-simvastatina en comparación con la simvastatina sola.Características en grupos especiales de pacientes: Niños: La absorción y el metabolismo de la ezetimiba son similares en los niños, los adolescentes (10 a 18 años) y los adultos. Basándose en la ezetimiba total, no hay diferencias farmacocinéticas entre los adolescentes y los adultos. No hay datos farmacocinéticos disponibles en niños menores de diez años.Pacientes de edad avanzada: Las concentraciones plasmáticas de ezetimiba total son aproximadamente el doble en los pacientes de edad avanzada (mayores de 65 años) que en los pacientes jóvenes (de 18 a 45 años).La disminución del C-LDL y el perfil de seguridad son similares en los de edad avanzada y en los jóvenes tratados con ezetimiba. Insuficiencia hepática: Después de una sola dosis de 10 mg de ezetimiba, el promedio del ABC de ezetimiba total fue aproximadamente 1.7 veces mayor en los pacientes con insuficiencia hepática leve (puntuación de Child-Pugh de 5 o 6) que en los sujetos sanos. En un estudio con dosis múltiples (10 mg diarios) durante 14 días en pacientes con insuficiencia hepática moderada (puntuación de Child-Pugh de 7 a 9), el promedio del ABC de ezetimiba total fue aproximadamente cuatro veces mayor los días 1 y 14 que en los sujetos sanos. No es necesario ajustar la dosificación en los pacientes con insuficiencia hepática leve.Debido a que se desconocen los efectos de la exposición aumentada de la ezetimiba en los pacientes con insuficiencia hepática moderada o intensa (puntuación de Child-Pugh mayor de 9), no se recomienda tratar con ezetimiba a esos pacientes (véase Precauciones generales). Insuficiencia renal: Ezetimiba: En ocho pacientes con enfermedad renal intensa (promedio de depuración de la creatinina ≥ 30 ml/min/1.73 m2), después de una sola dosis de 10 mg de ezetimiba el promedio de ABC de ezetimiba total fue aproximadamente 1.5 veces mayor que en nueve sujetos sanos.En otro paciente de ese estudio (con trasplante renal y recibiendo varios medicamentos, incluyendo ciclosporina), la exposición a la ezetimiba total fue 12 veces mayor.Simvastatina: En un estudio en pacientes con insuficiencia renal intensa (depuración de la creatinina < 30 ml/min), las concentraciones plasmáticas de inhibidores totales después de una sola dosis de un inhibidor de la reductasa de la HMG-CoA relacionado con la simvastatina fueron aproximadamente el doble que en los voluntarios sanos. Sexo: Las concentraciones plasmáticas de ezetimiba total son ligeramente mayores (menos de 20%) en las mujeres que en los hombres. La disminución del C-LDL y el perfil de seguridad son similares en los hombres y en las mujeres tratados con ezetimiba.Raza: Basándose en un meta-análisis de estudios farmacocinéticos con ezetimiba, no hubo diferencias farmacocinéticas entre las personas de raza negra y las de raza blanca.
Contraindicaciones: Hipersensibilidad a cualquiera de los componentes de este producto.Enfermedad hepática activa o aumento persistente inexplicable de las transaminasas séricas.Embarazo y lactancia (véase Restricciones de uso durante el embarazo y la lactancia).Administración concomitante de gemfibrozilo, ciclosporina o danaza. La administración concomitante de inhibidores potentes de CYP3A4, inhibidores de la proteasa del VIH, boceprevir, teleprevir, eritromicina, claritromicina, telitromicina, nefazodona y los fármacos que contienen cobicistat.
Precauciones generales: Cuando se administre fenofibrato con ezetimiba/simvastatina consulte la información para prescribir de fenofibrato. Miopatía/rabdomiólisis: Como otros inhibidores de la reductasa de la hidroximetilglutaril-coenzima A (HMGCoA), la simvastatina causa ocasionalmente miopatía, manifestada por dolor, hiperestesia o debilidad muscular y aumento de la cinasa de la creatina a más de diez veces el límite normal superior. En algunos casos la miopatía toma la forma de rabdomiólisis, con o sin insuficiencia renal aguda secundaria a mioglobulinuria, y en raros casos ha fallecido el paciente. Una gran actividad inhibidora de la reductasa de la HMG-CoA en el plasma aumenta el riesgo de miopatía. Entre los factores de predisposición para miopatía se incluyen edad avanzada (3 65 años), género femenino, hipotiroidismo incontrolado y deficiencia renal.En un ensayo clínico en el cual los pacientes con un antecedente de infarto al miocardio fueron tratados con simvastatina 80 mg/día (media de seguimiento 6.7 años), la incidencia de miopatía fue aproximadamente 1.0% en comparación con 0.02% para los pacientes que recibieron 20 mg/día. Aproximadamente la mitad de estos casos con miopatía se produjo durante el primer año de tratamiento. La incidencia de miopatía durante cada año subsiguiente de tratamiento fue aproximadamente de 0.1%.Debido a que ezetimiba/simvastatina contiene simvastatina, el riesgo de miopatía/rabdomiólisis aumenta por el uso concomitante de ezetimiba/simvastatina con:1. Se debe evitar el uso concomitante de ezetimiba/simvastatina con inhibidores potentes de CYP3A4 (por ejemplo, itraconazol, ketoconazol, eritromicina, claritromicina, telitromicina, inhibidores de la proteasa del VIH o nefazodona). Si el tratamiento con itraconazol, ketoconazol, eritromicina, claritromicina o telitromicina es inevitable, se debe suspender la administración de ezetimiba/simvastatina durante él. Se debe evitar el uso concomitante de otros medicamentos que a dosis terapéuticas tengan un potente efecto inhibidor de la CYP3A4 a menos que los beneficios del tratamiento combinado justifiquen el aumento del riesgo (véase Interacciones medicamentosas y de otro género). 2. Con relación a los fibratos, sólo se ha estudiado la seguridad y eficacia de ezetimiba/simvastatina administrado con fenofibrato. Debido a lo anterior el uso concomitante de ezetimiba/simvastatina y fibratos, excepto fenofibrato, se debe evitar (véase Interacciones medicamentosas y de otro genero). No se han estudiado dosis mayores a 10/20 mg/día de ezetimiba/simvastatina y fenofibrato. Se debe tener precaución cuando se prescriba ezetimiba/simvastatina y fenofibrato, debido a que el fenofibrato como monoterapia puede causar miopatía. En un estudio de 12 semanas en el cual 184 pacientes recibieron 10/20 mg/día de ezetimiba/simvastatina más 160 mg/día de fenofibrato, la coadministración fue bien tolerada. En otro estudio de 12 semanas, en el cual 411 pacientes recibieron 20 mg/día de simvastatina y 160 mg/día de fenofibrato, la coadministración también fue bien tolerada. Existe un aumento en el riesgo de miopatía cuando se usa simvastatina concomitantemente con fibratos (especialmente gemfibrozil). El uso combinado de simvastatina con gemfibrozil se debe evitar, a menos que los beneficios puedan superar los riesgos de esta combinación. La dosis de simvastatina no debe xceder los 10 mg diarios en pacientes recibiendo tratamiento concomitante con gemfibrozil. Por lo tanto, aunque no es recomendable, si ezetimiba/simvastatina se utiliza en combinación con gemfibrozil, la dosis no debe exceder de 10/10 mg diarios.3. En los pacientes bajo tratamiento concomitante con ezetimiba/simvastatina y ciclosporina o danazol, la dosificación de ezetimiba/simvastatina no debe ser mayor de 10/10 mg al día. Los beneficios con el empleo de ezetimiba/simvastatina en pacientes que reciben ciclosporina o danazol se deben de valorar cuidadosamente contra los riesgos que pueden presentarse con esta combinación y se debe tener precaución al empezar a administrar ezetimiba/simvastatina a un paciente que ya está tomando ciclosporina (véase Interacciones medicamentosas y de otro género).4. En los pacientes bajo tratamiento concomitante con ezetimiba/simvastatina y amiodarona o verapamil, la dosificación de ezetimiba/simvastatina no debe ser mayor de 10/20 mg al día. En un ensayo clínico, se reportó miopatía en 6% de los pacientes que recibieron 80 mg diarios de simvastatina y amiodarona. Se debe evitar el uso combinado de más de 10/20 mg diarios de ezetimiba/simvastatina y amiodarona o verapamil, a menos que sea muy probable que los beneficios justifiquen el aumento del riesgo de miopatía (véase Interacciones medicamentosas y de otro género).5. La dosis de ezetimiba/simvastatina no debe exceder 10/40 mg diarios en pacientes que reciben medicación concomitante con diltiazem, a menos que el posible beneficio clínico pese más que el aumento del riesgo de miopatía. En los pacientes que están tomando diltiazem y reciben simvastatina 80 mg tuvieron un aumentado riesgo de miopatía. En los estudios clínicos, el riesgo de miopatía fue similar en los pacientes que recibieron 40 mg diarios de simvastatina y diltiazem y en los que recibieron sólo los 40 mg diarios de simvastatina (véase Interacciones medicamentosas y de otro género).6. Se debe tener precaución cuando se prescriba amlodipina con ezetimiba/simvastatina 10/80 mg ya que hay un ligero aumento en el riesgo de miopatía con el uso concomitante. En un estudio clínico con pacientes que recibieron amlodipina concomitantemente con simvastatina 80 mg tuvieron ligeramente aumentado el riesgo de miopatía. El riesgo de miopatía en pacientes que recibieron simvastatina 40 mg no fue aumentado al tomar amlodipina concomitantemente (véase Interacciones medicamentosas y de otro género).7. Se han observado casos de miopatía/rabdomiólisis con simvastatina coadministrada con niacina (dosis ≤ 1 g/día), modificador de lípidos. En un estudio en curso, doble-ciego, con distribución al azar, llevado a cabo en China, Reino Unido y Escandinavia. Un análisis provisional de los resultados cardiovasculares hecho por un comité independiente de monitoreo de la seguridad reveló que la incidencia de la miopatía entre aproximadamente 4,700 pacientes UK/escandinavo tratados con simvastatina 40 mg o ezetimiba/simvastatina 10/40 mg coadministrados con niacina/laropiprant 2 g/40 mg de liberación prolongada (ER) es similar a la incidencia global informada en la base de datos del ensayo clínico para simvastatina 40 mg (0.08%). Sin embargo, en aproximadamente 3,900 pacientes chinos en el mismo brazo de tratamiento, la incidencia es mayor de la esperada (aproximadamente el 0.9%). No aumentó el riesgo de miopatía entre los 8,600 pacientes chinos, ingleses o escandinavos en el brazo de control (placebo más simvastatina 40 mg o ezetimiba/simvastatina 10/40 mg). No hubo ninguna contribución aparente de ezetimiba a la incidencia aumentada de miopatía. Debido a que la incidencia de miopatía es superior en los pacientes chinos que en los pacientes no-chinos, se debe tener precaución en el tratamiento de pacientes chinos con ezetimiba/simvastatina (particularmente dosis de 10/40 mg o superiores) coadministradas con el modificador de lípidos, niacina a dosis ≤ 1 g/día o productos que contienen niacina. Debido a que el nesgo de miopatía está relacionado con la dosis, no se recomienda el uso de ezetimiba/simvastatina 10/80 mg con el modificador de lípidos niacina (dosis ≤ 1 g/día) o con productos que contienen niacina en pacientes chinos. Se desconoce si hay un incremento en el riesgo de miopatía con la coadministración en otros pacientes asiáticos.8. Los pacientes en tratamiento con ácido fusídico y ezetimiba/simvastatina deben vigilarse estrechamente. Debe de considerarse la suspensión temporal del tratamiento con ezetimiba/simvastatina.9. Al iniciar el tratamiento con ezetimiba/simvastatina o al aumentar su dosificación se les debe informar a todos los pacientes sobre el riesgo de miopatía e indicarles que reporten enseguida cualquier dolor, hiperestesia o debilidad musculares. Si se diagnostica o se sospecha miopatía, se debe suspender inmediatamente el tratamiento con ezetimiba/simvastatina. Esos síntomas y/o una concentración de creatina cinasa más de diez veces mayor que el límite superior de la normal indican miopatía. En la mayoría de los casos, cuando se suspendió inmediatamente el tratamiento con simvastatina cesaron los síntomas musculares y el aumento de la cinasa de la creatina. Se puede considerar medir periódicamente la cinasa de la creatina en los pacientes que empiezan a tomar ezetimiba/simvastatina o en los que se aumenta la dosificación. Se recomiendan las determinaciones periódicas de creatina cinasa para pacientes con ajuste de dosis a 10/80 mg. No se puede asegurar que esa vigilancia evitará la miopatía.10.Muchos de los pacientes que han presentado rabdomiólisis durante el tratamiento con simvastatina tenían complicaciones médicas previas, incluyendo insuficiencia renal debida usualmente a diabetes mellitus de larga duración. Esos pacientes requieren una vigilancia más estrecha. Se debe suspender temporalmente el tratamiento con ezetimiba/simvastatina unos cuantos días antes de una intervención de cirugía mayor y cuando surge cualquier trastorno médico o quirúrgico importante.Enzimas hepáticas: En ensayos comparativos en pacientes a los que se les coadministraron ezetimiba y simvastatina se han observdo aumentos sucesivos de las transaminasas (al triple o más del límite superior de sus valores normales) (vease Reacciones secundarias y adversas).Se recomienda hacer pruebas del funcionamiento hepático antes de iniciar el tratamiento con ezetimiba/simvastatina y después cuando esté clínicamente indicado. En los pacientes en los que se aumente la dosificación hasta 10/80 mg diarios se deben repetir las pruebas del funcionamiento hepático antes del aumento, a los tres meses de éste, y después periódicamente (por ejemplo, cada seis meses) durante un año. Se debe prestar especial atención a los pacientes que presenten aumentos de las concentraciones de las transaminasas séricas; en ellos, las mediciones se deben repetir pronto y con mayor frecuencia.Si las concentraciones de las transaminasas siguen aumentando, particularmente si llegan al triple del límite normal superior y son persistentes, se debe suspender la administración del medicamento. Ezetimiba/simvastatina se debe usar con precaución en los pacientes que toman cantidades considerables de alcohol o que tienen antecedentes de enfermedad hepática. Las enfermedades hepáticas activas y los aumentos persistentes de las transaminasas son contraindicaciones para el uso de ezetimiba/simvastatina. Insuficiencia hepática: Debido a que se desconocen los efectos de la exposición aumentada a la ezetimiba en los pacientes con insuficiencia hepática moderada o intensa, no se recomienda tratar con ezetimiba/simvastatina a esos pacientes (véase Farmacocinética y farmacodinamia, Características en grupos especiales de pacientes).Fibratos: Con relación a los fibratos, sólo se ha estudiado la seguridad y eficacia de ezetimiba/simvastatina administrado con fenofibrato. Debido a lo anterior, el uso concomitante de ezetimiba/simvastatina y fibratos, excepto fenofibrato, se debe evitar (véase Interacciones medicamentosas y de otro género).Fenofibrato: Si se sospecha la presencia de colelitiasis en un paciente que esté tomando ezetimiba/simvastatina y fenofibrato, se recomienda la realización de estudios de la vesícula biliar y se debe considerar otro tratamiento reductor de lípidos (véase Reacciones secundarias y adversas y la información para prescribir de fenofibrato).Ciclosporina: Se debe tener precaución al iniciar ezetimiba/simvastatina en pacientes que ya estén tomando ciclosporina. Las concentraciones de ciclosporina deben vigilararse en los pacientes que reciben ezetimiba/simvastatina y ciclosporina (véase Interacciones medicamentosas y de otro género).Anticoagulantes: Si ezetimiba/simvastatina se agrega a la warfarina otro anticoagulante cumarínico, o fluindiona, la proporción normalizada internacional (PNI) debe ser apropiadamente vigilada (véase Interacciones medicamentosas y de otro género).Uso pediátrico: Se evaluó la seguridad y la eficacia de ezetimiba/simvastatina en pacientes 10 a 17 años de la edad con hipercolesterolemia familiar heterocigótica en un ensayo clínico controlado en hombres y mujeres que eran por lo menos un año posmenarca. Los pacientes adolescentes tratados con ezetimiba/simvastatina tenían un perfil de una experiencia adversa similar a la de los pacientes adultos tratados con ezetimiba/simvastatina. No se han estudiado dosis superiores a 10/40 mg/día en esta población.En este estudio controlado, no hubo efecto detectable sobre crecimiento o la maduración sexual de los adolescentes, o algún efecto sobre longitud del ciclo menstrual en las adolescentes (véase Dosis y vía de administración y Reacciones secundarias y adversas), ezetimiba/simvastatina no se ha estudado en pacientes más jóvenes de 10 años de edad o en jóvenes premenarcas.Uso en pacientes de edad avanzada: Debido a que la edad avanzada (≤ 65 años) es un factor de predisposición para miopatía, ezetimiba/simvastatina debe ser prescrito con precaución en las personas de edad avanzada. En un ensayo clínico de pacientes tratados con simvastatina 80 mg/día, los pacientes ≤ 65 años de edad tuvieron un mayor riesgo de miopatía en comparación con los pacientes de 65 años de edad.Enfermedad pulmonar intersticial: Se han notificado casos de enfermedad pulmonar intersticial con algunas estatinas, incluida la simvastatina, especialmente con terapia a largo plazo. Las características de presentación pueden incluir disnea, tos no productiva y deterioro en la salud general (fatiga, pérdida de peso y fiebre). Si se sospecha que un paciente ha desarrollado enfermedad pulmonar intersticial, se debe suspender el tratamiento con ezetimiba/simvastatina.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: La aterosclerosis es un proceso crónico, y ordinariamente suspender la administración de medicamentos reductores de los lípidos durante el embarazo debe tener poco efecto sobre el riesgo a largo plazo asociado con la hipercolesterolemia primaria.Ezetimiba/simvastatina está contraindicado durante el embarazo (véase Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad, Desarrollo tetal).Simvastatina: No se ha determinado la seguridad de la simvastatina en mujeres embarazadas, ni se han hecho ensayos clínicos comparativos con simvastatina en ellas. Se han recibido escasos reportes de anomalías congénitas tras la exposición intrauterina a inhibidores de la reductasa de la HMG-CoA, pero en un estudio prospectivo de unos 200 embarazos en los que hubo exposición del feto durante el primer trimestre a la simvastatina o a otro inhibidor de la reductasa de la HMG-CoA muy relacionado con la simvastatina, la incidencia de anomalías congénitas fue similar a la observada en la población general.Ese número de embarazos fue estadísticamente suficiente para excluir un aumento de las anomalías congénitas de 2.5 veces o más sobre su incidencia en la población general.Aunque no hay ningún indicio de que la incidencia de anomalías congénitas en los hijos de pacientes que han tomado simvastatina u otro inhibidor de la reductasa de la HMG-CoA muy relacionado con ella difiera de la observada en la población general, el tratamiento de las embarazadas con simvastatina puede disminuir las concentraciones fetales de mevalonato, que es un precursor en la síntesis de colesterol. Por lo tanto, no se debe usar ezetimiba/simvastatina en mujeres embarazadas, que están tratando de embarazarse o que pueden estar embarazadas.El tratamiento con ezetimiba/simvastatina se debe suspender durante todo el embarazo o hasta que se haya comprobado que la paciente no esta embarazada (vease Contraindicaciones).Ezetimiba: No hay datos clínicos sobre la administración de ezetimiba a mujeres embarazadas. Cuando se administró ezetimiba con simvastatina a ratas embarazadas en estudios sobre el desarrollo embriofetal, no se observó ningún efecto teratogénico. En conejas embarazadas hubo una baja incidencia de malformaciones esqueléticas (véase Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad, Desarrollo fetal). Madres lactantes: Los estudios en ratas han mostrado que la ezetimiba es excretada con la leche.No se sabe si los componentes activos de ezetimiba/simvastatina son excretados con la leche humana, por lo que no se debe administrar ezetimiba/simvastatina a mujeres que están amamantando.
Reacciones secundarias y adversas: Se ha evaluado la seguridad de Ezetimiba/Simvastatina (o la coadministración de ezetimiba y simvastatina equivalente a las formulaciones) en ensayos clínicos sobre aproximadamente 12,000 pacientes, y Ezetimiba/Simvastatina fue generalmente bien tolerado. En los pacientes que tomaron Ezetimiba/Simvastatina (n = 2404) se reportaron las siguientes reacciones adversas; relacionadas con el medicamento y en una mayor incidencia que aquellos a los que se les administró placebo (N = 1340). Las siguientes se presentan por frecuencia (MedDRA, 2004), y las definiciones utilizadas son: Muy común (≥ 1/10) Común (≥ 1/100, < 1/10), poco común (≥ 1/1,000, < 1/100), rara (≥ 1/10,000, < 1/1,000); muy rara vez ( < 1/10,000), Caso Aislado( < 3 casos) o Frecuencia desconocida: no puede calcularse con los datos disponibles: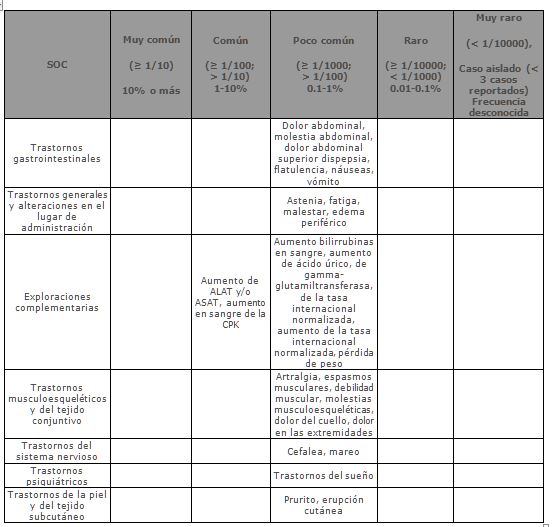
En los pacientes que tomaron Ezetimiba/Simvastatina (n=9595) se reportaron las siguientes reacciones adversas; relacionadas con el medicamento y en una mayor incidencia que aquellos que se les administró una estatina sola (N=8883).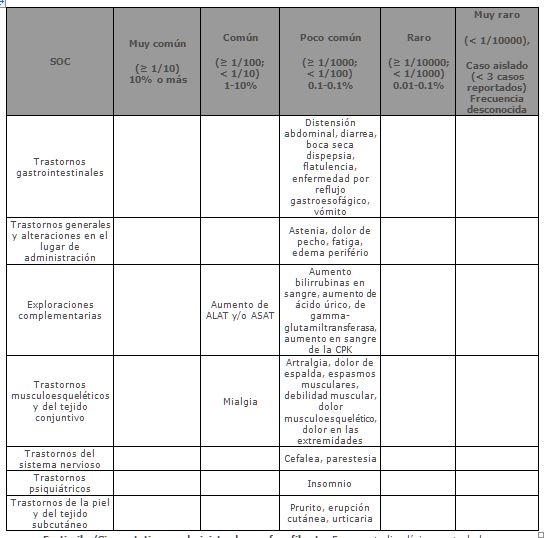
Ezetimiba/Simvastatina coadministrado con fenofibrato: En un estudio clínico controlado, el perfil de eventos adversos reportado para la coadministración de Ezetimiba/Simvastatina y fenofibrato fue consistente con los perfiles reportados para Ezetimiba/Simvastatina y/o fenofibrato solos.Pacientes pediátricos (10 a 17 años de edad): En un estudio con pacientes adolescentes (10 a 17 años de edad) con hipercolesterolemia familiar heterocigótica (n=248), el perfil de la seguridad y la tolerabilidad del grupo tratado con Ezetimiba/Simvastatina fueron similares al de los pacientes adultos tratados con Ezetimiba/Simvastatina.Experiencia después de la comercialización: Las siguientes reacciones adversas adicionales han sido reportadas después de la comercialización de ezetimiba/simvastatina o durante los estudios clínicos o durante el uso después de la comercialización de cualquiera de los componentes individuales de este producto. Las reacciones adversas reportadas para ezetimiba/simvastatina son consistentes con aquellas reportadas previamente con ezetimiba y/o simvastatina.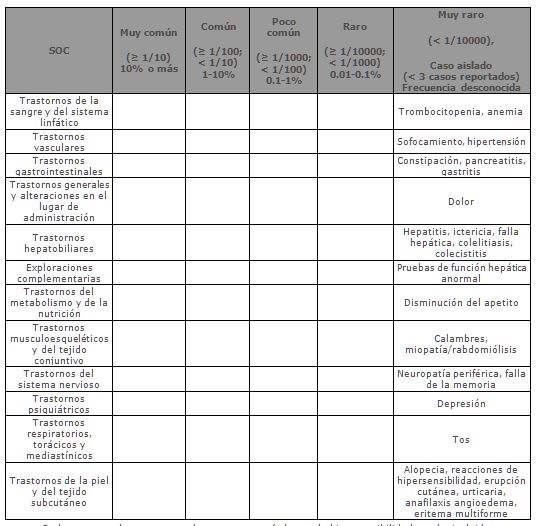
Se han reportado casos raros de un aparente síndrome de hipersensibilidad que ha incluido algunas de las siguientes alteraciones: Edema angioneurótico, síndrome tipo lupus, polimialgia reumática, dermatomiositis, vasculitis, trombocitopenia, eosinofilia, aumento de la velocidad de sedimentación eritrocítica, artritis y artralgia, urticaria, fotosensibilidad, fiebre, rubefacción, disnea y malestar general.Ezetimiba coadministrada con fenofibrato: En un estudio clínico multicéntrico, doble-ciego, controlado con placebo, en pacientes con hiperlipidemia mixta, se dio tratamiento a 625 pacientes hasta por 12 semanas y a 576 hasta por 1 año. Este estudio no fue diseñado para comparar grupos de tratamiento en búsqueda de eventos poco frecuentes.Las tasas de incidencia (IC 95%) para aumentos clínicamente importantes ( > 3 x LSN, consecutivos) en las transaminasas séricas fue de 4.5% (1.9, 8.8) y 2.7% (1.2, 5.4) para la monoterapia con fenofibrato y ezetimiba coadministrada con fenofibrato, respectivamente, ajustada para la exposición al tratamiento. Las tasas de incidencia correspondientes para colecistectomía fueron de 0.6% (0.0, 3.1) y 1.7% (0.6, 4.0) para la monoterapia con fenofibrato y ezetimiba coadministrada con fenofibrato, respectivamente (véase Precauciones generales). No hubo aumentos de la CPK > 10 x LSN en ninguno de los grupos de tratamiento en este estudio.
Interacciones medicamentosas y de otro género: Ezetimiba/simvastatina: No se observó ninguna interacción farmacocinética de importancia clínica cuando se coadministraron ezetimiba y simvastatina.EXOTIB DUO® es bioequivalente a la coadministración de ezetimiba y simvastatina.Interacciones con la enzima CYP3A4: Los estudios preclínicos han mostrado que la ezetimiba no induce las enzimas metabolizadoras de medicamentos del citocromo P-450.No se ha observado ninguna interacción de importancia clínica entre la ezetimiba y medicamentos que son metabolizados por los citocromos P-450 1A2, 206, 2C8, 2C9 y 3A4 o por la acetiltransferasa.La simvastatina es metabolizada por la CYP3A4, pero no inhibe su actividad; por lo tanto, no es de esperarse que afecte las concentraciones plasmáticas de otros medicamentos metabolizados por la CYP3A4.Los siguientes inhibidores potentes de la CYP3A4 aumentan el riesgo de miopatía al disminuir la eliminación del componente simvastatina de ezetimiba/simvastatina (véase Precauciones generales, Miopatía/rabdomiólisis): ltraconazol,Ketoconazol,Eritromicina,Claritromicina,Telitromicina,lnhibidores de la proteasa del VIH,Nefazodona.Interacciones con medicamentos reductores de los lípidos que pueden causar miopatía cuando se administran solos: También aumentan el riesgo de miopatía los siguientes medicamentos reductores de los lípidos que no son inhibidores potentes de la CYP3A4, pero pueden causar miopatía cuando se administran solos (véase Precauciones generales, Miopatía/rabdomiólisis): Gemfibrozil. Otros fibratos.En un estudio, en el cual se coadministraron 10/20 mg/día de ezetimiva/simvastatina y 160 mg/día de fenofibrato en 184 pacientes hasta por 12 semanas, no hubo reportes de miopatía. Interacciones con otros medicamentos: Ciclosporina o danazol: El riesgo de miopatía/rabdomiólisis se incrementa con la administración concomitante de ciclosporina o danazol, particularmente con dosis altas de ezetimiba/simvastatina (véase Precauciones generales, Miopatía/rabdomiólisis). Amiodarona: El riesgo de miopatía/rabdomiólisis aumenta cuando se coadministran amiodarona con dosis altas de ezetimiba/simvastatina (véase Precauciones generales, Miopatía/rabdomiólisis).Colestiramina: La administración concomitante de colestiramina disminuyó 55% aproximadamente el promedio del área bajo la curva (ABC) de concentración de la ezetimiba total (ezetimiba + glucurónido de ezetimiba). Esa interacción puede hacer que sea menor la disminución adicional del C-LDL debido a la coadministración de ezetimiba/simvastatina y colestiramina. Bloqueadores de canales de calcio: Verapamil: La administración concomitante de verapamil con dosis altas de ezetimiba/simvastatina aumenta el riesgo de miopatía/rabdomiólisis (véase Precauciones generales, Miopatía/rabdomiólisis). Diltiazem: El riesgo de miopatía/rabdomiólisis aumenta por la administración concomitante de diltiazem y ezetimiba/simvastatina 10/80 (véase Precauciones generales, Miopatía/rabdomiólisis).Amlodipina: Los pacientes que recibieron tratamiento con amlodipina concomitantemente con ezetimiba/simvastatina 10/80 mg tuvieron ligeramente aumentado el riesgo de miopatía (véase Precauciones generales, Miopatía/rabdomiólisis).Ácido fusídico: Los pacientes en tratamiento con ácido fusídico que reciben tratamiento concomitante con ezetimiba/simvastatina pueden tener un riesgo incrementado de miopatía (véase Precauciones generales, Miopatía/rabdomiólisis).Fibratos: Con relación a los fibratos, sólo se ha estudiado la seguridad y eficacia de ezetimiba/simvastatina administrado con fenofibrato.En un estudio, en el cual se coadministraron 10/20 mg/día de Ezetimiba/simvastatina y 160 mg/día de fenofibrato en 184 pacientes hasta por 12 semanas, ningún paciente experimentó eventos relacionados con la vesícula biliar. Los fibratos pueden aumentar la excreción de colesterol con la bilis y producir así colelitiasis.La seguridad y eficacia de ezetimiba coadministrada con fenofibrato ha sido evaluada en un estudio clínico (véase Reacciones secundarias y adversas); no se ha estudiado la coadministración de ezetimiba con otros fibratos.En un estudio preclínico en perros, la ezetimiba aumentó el contenido de colesterol de la bilis. Aunque se desconoce la importancia de ese resultado preclínico para los seres humanos, no se recomienda la coadministración de ezetimiba/simvastatina y fibratos, que no sea fenofibrato, hasta que se estudie su uso en pacientes.Fenofibrato: En un estudio de farmacocinética, la administración concomitante de fenofibrato aumentó las concentraciones totales de ezetimiba aproximadamente 1.5 veces. Este aumento no se considera como clínicamente significativo.Gemfibrozil: En un estudio de farmacocinética, la administración concomitante, de gemfibrazil aumentó las concentraciones totales de ezetimiba aproximadamente 1.7 veces.Este aumento no se considera como clínicamente significativo. No están disponibles datos clínicos.Niacina: En un estudio de 15 adultos sanos, ezetimiba/simvastatina (10/20 mg diarios por 7 días) causó un incremento mínimo en la media del ABC de niacina (22%) y del ácido nicotinúrico (19%) cuando se administró concomitantemente con tabletas de niacina de liberación prolongada (1,000 mg por 2 días y 2,000 mg por 5 días seguido de un desayuno bajo en grasa). En el mismo estudio, la administración de niacina concomitante aumentó ligeramente la media del ABC de ezetimiba (9%), de ezetimiba total (26%), de simvastatina (20%) y del ácido del simvastatina (35%). Estos incrementos no se considerar clínicamente significativos (véase Precauciones generales, Miopatíalrabdomiólisis).Otras interacciones: El jugo de toronja contiene uno o más componentes que inhiben la CYP3A4 y pueden aumentar las concentraciones plasmáticas de los medicamentos metabolizados por la CYP3A4.El efecto del consumo típico de jugo de toronja (250 ml al día) es mínimo (aumento de 13% de la actividad inhibidora de la reductasa de la HMG-CoA en el plasma, medida por el ABC) y no tiene ninguna importancia clínica. Sin embargo, las cantidades muy grandes de jugo de toronja (más de un litro al día) aumentan significativamente la actividad inhibidora de la reductasa de la HMG-CoA en el plasma, por lo que se deben evitar durante el tratamiento con ezetimiba/simvastatina (véase Precauciones generales, Miopatíalrabdomiólisis). Anticoagulantes: En dos estudios clínicos, uno en voluntarios sanos y el otro en pacientes hipercolesterolémicos, 20-40 mg diarios de simvastatina potenciaron ligeramente el efecto de anticoagulantes cumarínicos. El tiempo de protrombina, reportado como la Razón Normalizada Internacional, aumentó sus valores iniciales de 1.7 a 1.8 en los voluntarios sanos y de 2.6 a 3.4 en los pacientes con hipercolesterolemia.En los pacientes que están tomando anticoagulantes cumarínicos se debe determinar el tiempo de protrombina antes de iniciar el tratamiento con ezetimiba/simvastatina y con suficiente frecuencia durante el principio del tratamiento para asegurarse de que no ocurra ninguna alteración significativa del tiempo de protrombina.Una vez que se haya comprobado que el tiempo de protrombina es estable, se puede seguir vigilando a los intervalos usualmente recomendados en los pacientes tratados con anticoagulantes cumarínicos. Si se cambia la dosificación o se suspende la administración de ezetimiba/simvastatina se debe repetir ese mismo procedimiento. El tratamiento con simvastatina no se ha asociado con sangrado o con cambios del tiempo de protrombina en pacientes que no están tomando anticoagulantes.En un estudio clínico de 12 hombres adultos sanos, la administración concomitante de ezetimiba (10 mg una vez al día) no tuvo un efecto significativo en la biodisponibilidad del tiempo de warfarina o protrombina. Ha habido reportes después de la comercialización de incrementos en la Proporción Normalizada Internacional en pacientes que han agregado ezetimiba/simvastatina a warfarina o fluinidiona. La mayoría de estos pacientes también tomaban otros medicamentos (véase Precauciones generales).El efecto de ezetimiba/simvastatina en el tiempo de protrombina no ha sido estudiado.Antiácidos: La administración concomitante de antiácidos disminuyó la rapidez de absorción de la ezetimiba, pero no tuvo ningún efecto sobre su biodisponibilidad. Esa disminución de la rapidez de absorción de la ezetimiba no se considera clínicamente importante.Ciclosporina: En un estudio de ocho pacientes con trasplante renal que tenían una depuración de la creatinina mayor de 50 ml/min bajo una dosificación estable de ciclosporina, una sola dosis de 10 mg de ezetimiba aumentó 3.4 veces (rango, 2.3 a 7.9 veces) el promedio de ABC de ezetimiba total en comparación con un grupo testigo de personas sanas de otro estudio (n = 17). En otro estudio, un paciente con trasplante renal e insuficiencia renal intensa (depuración de la creatinina de 13.2 ml/min/1.73 m2) que estaba recibiendo múltiples medicamentos, incluyendo ciclosporina, tuvo una exposición a la ezetimiba total 12 veces mayor que los testigos.En un estudio de doce sujetos sanos de dos periodos, cruzado, la administración diaria de 20 mg de ezetimiba por 8 días con una dosis única de 100 mg de ciclosporina en el día 7 dio como resultado un incremento promedio de 15% en el ABC de la ciclosporina (rango de 10% aumento a 51% de disminución) comparado con una dosis única de ciclosporina sola (véase Precauciones generales).
Alteraciones en los resultados de pruebas de laboratorio: En ensayos clínicos comparativos sobre la coadministración de ezetimiba y simvastatina, la incidencia de aumentos clí