FORXIGA
ASTRAZENECA
Denominación genérica: Dapagliflozina.
Forma famacéutica y formulación: Tabletas. Cada tableta contiene: Dapaglifozina propanodiol equivalente a 5.0 mg o 10.0 mg de Dapaglifozina. Excipiente cbp.
Indicaciones terapéuticas: Monoterapia: FORXIGA® está indicado como complemento a la dieta y el ejercicio para mejorar el control glucémico en pacientes con diabetes mellitus tipo 2 (DMT2). Terapia de adición: FORXIGA® está indicado en pacientes con diabetes mellitus tipo 2 para mejorar el control glucémico en combinación con metformina, una tiazolidinediona, una sulfonilurea (con o sin metformina), un inhibidor de DPP4 (con o sin metformina), o insulina (sola o en combinación con hasta dos antidiabéticos orales) cuando la terapia existente, junto con dieta y ejercicio no proporcionan control glucémico adecuado. Combinación inicial: FORXIGA® está indicado para su uso como terapia de combinación inicial con metformina, como complemento a la dieta y el ejercicio, para mejorar el control glucémico en pacientes con diabetes mellitus tipo 2 cuando es apropiada la terapia dual de dapagliflozina y metformina.
Farmacocinética y farmacodinamia: FORXIGA® (dapagliflozina propanodiol) es un potente y altamente selectivo inhibidor oral del cotransportador de sodio-glucosa tipo 2 (SGLT2) en humanos, el principal transportador responsable de la reabsorción de glucosa renal. Farmacocinética: Absorción: Dapagliflozina se absorbe bien y rápidamente después de la administración oral y se puede administrar con o sin alimentos. Las concentraciones plasmáticas máximas de dapagliflozina (Cmáx.) se alcanzaron por lo general a las 2 horas de su administración en ayunas. Los valores de Cmáx. y exposición sistémica a dapagliflozina (AUC) se incrementaron en forma proporcional al incremento en la dosis de dapagliflozina. La biodisponibilidad oral absoluta de dapagliflozina, después de la administración de una dosis de 10 mg, es de 78%. Los alimentos tuvieron un efecto relativamente modesto en la farmacocinética de dapagliflozina en sujetos sanos. La administración con una comida alta en grasas disminuyó la Cmáx. de dapagliflozina en hasta 50% y prolongó el Tmáx. en aproximadamente 1 hora, pero no alteró el AUC en comparación con el estado en ayunas. Estos cambios no se consideran clínicamente significativos. Distribución: Dapagliflozina se une a proteínas aproximadamente en 91%. La unión a proteínas no se vio alterada en varios estados patológicos (por ejemplo, insuficiencia renal o hepática). Metabolismo: Dapagliflozina consta de un enlace glucosídico, lo que significa que el componente aglicona está unido a la glucosa mediante un enlace carbono-carbono, lo que le confiere estabilidad contra enzimas glucosidasas. La vida media terminal plasmática (t½) para dapagliflozina es de 12.9 horas después de una dosis oral única de FORXIGA® 10 mg en sujetos sanos. Dapagliflozina se metaboliza en forma extensa, principalmente para formar dapagliflozina 3-O-glucurónido, un metabolito inactivo. Dapagliflozina 3-O-glucurónido representó 61% de una dosis de 50 mg de (14C)-dapagliflozina, siendo el componente predominante relacionado con el fármaco en el plasma humano que representa 42% (basado en el AUC [0-12 h]) de la radiactividad plasmática total, similar a la contribución de 39% del fármaco original. Basado en el AUC, ningún otro metabolito representó > 5% de la radiactividad plasmática total en cualquier tiempo medido. Dapagliflozina 3-O-glucurónido u otros metabolitos no contribuyen a los efectos de disminución de la glucosa. La formación de dapagliflozina 3-O-glucurónido está medida por UGT1A9, una enzima presente en el hígado y el riñón, y el metabolismo mediado por CYP fue una vía de eliminación secundaria en humanos. Eliminación: Dapagliflozina y los metabolitos relacionados se eliminan principalmente a través de la excreción urinaria, de la cual menos de 2% es dapagliflozina intacta. Después de la administración de una dosis de 50 mg de (14C)-dapagliflozina, se recuperó 96, 75% en la orina y 21% en las heces. En heces, aproximadamente 15% de la dosis se excretó como fármaco original. Poblaciones especiales: No se recomiendan ajustes de dosis con base en análisis farmacocinéticos para insuficiencia renal leve a moderada, insuficiencia hepática leve, moderada y severa, edad, género, raza y peso corporal. Insuficiencia renal: FORXIGA® no debe ser usado en pacientes con insuficiencia renal moderada o severa (TFGe persistente < 45 mL/min/1.73 m2 o DepCr persistente < 60 mL/min) (véase Dosis y vía de administración). En estado estacionario (dapagliflozina 20 mg una vez al día durante 7 días), los pacientes con diabetes tipo 2 e insuficiencia renal leve, moderada y severa (determinado por depuración de iohexol), tuvieron exposiciones sistémicas promedio de dapagliflozina que fueron 32, 60 y 87% más altas, respectivamente, que en aquellos pacientes con diabetes tipo 2 y función renal normal. Con 20 mg de dapagliflozina una vez al día, la exposición sistémica más alta a dapagliflozina en pacientes con diabetes mellitus tipo 2 e insuficiencia renal, no resultó en una depuración renal más elevada de glucosa o excreción de glucosa en 24 horas. La depuración renal de glucosa y la excreción de glucosa de 24 horas fue inferior en pacientes con insuficiencia renal moderada o severa en comparación con pacientes con insuficiencia renal normal y leve. La excreción urinaria de glucosa en 24 horas fue altamente dependiente de la función renal, 85, 52, 18 y 11 g de glucosa/día se excretaron en pacientes con diabetes mellitus tipo 2 y función renal normal o insuficiencia renal leve, moderada o severa, respectivamente. No hubo diferencias en la unión a proteínas de dapagliflozina entre los grupos de insuficiencia renal o en comparación con sujetos sanos. Se desconoce el impacto de la hemodiálisis en la exposición a dapagliflozina. Insuficiencia hepática: Para recomendaciones de administración en pacientes con insuficiencia hepática moderada o severa, véase Dosis y vía de administración. Se realizó un estudio de farmacología clínica con dosis única de dapagliflozina (10 mg) en pacientes con insuficiencia hepática leve, moderada o severa (clases Child-Pugh A, B y C, respectivamente) y sujetos control sanos, con el fin de comparar las características farmacocinéticas de dapagliflozina entre estas poblaciones. No hubo diferencias en la unión a proteínas de dapagliflozina entre los grupos de pacientes con insuficiencia hepática en comparación con los sujetos sanos. En pacientes con insuficiencia hepática leve o moderada, la Cmáx. y el AUC promedio de dapagliflozina fue hasta de 12 y 36% superiores, respectivamente, en comparación con los sujetos control sanos. Estas diferencias no se consideraron clínicamente importantes y no se propone ajuste de dosis a partir de la dosis usual propuesta de 10 mg una vez al día de dapagliflozina para estas poblaciones. En pacientes con insuficiencia hepática severa (clase Child-Pugh C), la Cmáx. y el AUC promedio de dapagliflozina fueron de hasta 40 y 67% superiores, que en los sujetos control sanos, respectivamente. No se requiere ajuste de dosis en pacientes con insuficiencia hepática severa. Sin embargo, el riesgo-beneficio del uso de dapagliflozina en pacientes con insuficiencia hepática severa, se debería valorar en forma individual puesto que la seguridad y eficacia de dapagliflozina no se ha estudiado específicamente en esta población. Edad: No se recomienda ajuste de dosis de dapagliflozina a partir de la dosis de 10 mg una vez al día con base en la edad. El efecto de la edad (jóvenes: ≥ 18 a < 40 años [n = 105] y de edad avanzada: > 65 años [n = 224]) se evaluó como covariable en un modelo farmacocinético poblacional y se comparó en pacientes entre ≥40 a < 65 años utilizando los datos de estudios con sujetos sanos y pacientes. La exposición sistémica promedio a dapaglifloxina (AUC) en pacientes jóvenes se estima que fue 10.4% más baja que en el grupo de referencia (IC 90%: 87.9, 92.2%) y 25% más elevada en pacientes de la tercera edad en comparación con el grupo de referencia (IC 90%: 123, 129%). Estas diferencias en exposición sistémica no se consideraron de importancia clínica. Pacientes pediátricos y adolescentes: No se ha estudiado la farmacocinética en la población pediátrica y adolescente. Género: No se recomienda ajuste de dosis para dapagliflozina a partir de la dosis de 10 mg una vez al día con base en el género. El género se evaluó como covariable en un modelo farmacocinético poblacional mediante datos de estudios con sujetos sanos y pacientes. El AUC promedio de dapagliflozina en mujeres (n = 619) se estima 22% más elevada que en hombres (n = 634) (IC 90%: 117, 124). Raza: No se recomienda ajuste de dosis para dapagliflozina a partir de la dosis de 10 mg una vez al día con base en la raza. La raza (caucásica, negra o asiática) se evaluó como covariable en un modelo farmacocinético poblacional mediante datos de estudios con sujetos sanos y pacientes. Las diferencias en exposiciones sistémicas entre estas razas fueron pequeñas. En comparación con sujetos blancos (n = 1,147), los asiáticos (n = 47) no presentaron diferencia en las exposiciones sistémicas medias a dapagliflozina (IC 90%: 3.7% más bajas, 1% mas altas). En comparación con sujetos blancos, los sujetos negros (n = 43) presentaron exposiciones sistémicas medias a dapagliflozina estimadas de 4.9% más bajas (IC 90%: 7.7% más bajas, 3.7% más bajas). Peso corporal: No se recomienda ajuste de dosis para dapagliflozina a partir de la dosis propuesta de 10 mg una vez al día con base en el peso. En un análisis farmacocinético poblacional con datos de estudios con sujetos sanos y pacientes, las exposiciones sistémicas en sujetos con alto peso corporal (≥ 120 kg, n = 91) se estimaron en 78.3% (IC 90%: 78.2, 83.2%) de los de sujetos de referencia con peso corporal entre 75 y 100 kg. La diferencia se considera como pequeña; por lo tanto, no se recomienda ajuste de dosis a partir de la dosis propuesta de 10 mg de dapagliflozina una vez al día en pacientes con diabetes mellitus tipo 2 con alto peso corporal (≥120 kg). Los sujetos con bajo peso corporal ( < 50 kg) no se representaron bien en los estudios con sujetos sanos y pacientes usados en el análisis farmacocinético poblacional. Por lo tanto, las exposiciones sistémicas a dapagliflozina se simularon con un gran número de sujetos. Las exposiciones sistémicas simuladas medias a dapagliflozina en sujetos de bajo peso corporal se estimaron como 29% más elevadas que en sujetos con el peso corporal del grupo de referencia. La diferencia se considera como pequeña, y con base en estos resultados, no se recomienda ajuste de dosis a partir de la dosis propuesta de 10 mg de dapagliflozina una vez al día en pacientes con diabetes mellitus tipo 2 con bajo peso corporal ( < 50 kg). Farmacodinamia: General: Se observaron incrementos en la cantidad de glucosa excretada en la orina en sujetos sanos y en pacientes con diabetes mellitus tipo 2 seguidos de la administración de dapagliflozina (figura 1). Se excretó aproximadamente 70 g de glucosa en la orina por día (correspondiente a 280 kcal/día) a una dosis de dapagliflozina de 10 mg/día en pacientes con diabetes mellitus tipo 2 durante 12 semanas. Esta tasa de eliminación de glucosa alcanzó la excreción máxima de glucosa observada con una dosis de dapagliflozina de 20 mg/día. Se observó evidencia de excreción sostenida de glucosa en pacientes con diabetes mellitus tipo 2 que recibieron dapagliflozina 10 mg/día hasta por 2 años. Esta excreción urinaria de glucosa con dapagliflozina también resulta en diuresis osmótica e incrementos en el volumen urinario. El incremento en el volumen urinario en pacientes con diabetes mellitus tipo 2 tratados con FORXIGA® 10 mg fueron sostenidos a las 12 semanas y representaron aproximadamente 375 mL/día. El incremento en el volumen urinario se asoció con un aumento pequeño y transitorio en la excreción urinaria de sodio que no se asoció con cambios en las concentraciones séricas de sodio. La excreción urinaria de ácido úrico también se incrementó en forma transitoria (durante 3-7 días) y estuvo acompañada por una reducción en la concentración sérica de ácido úrico. A las 24 semanas, las reducciones en las concentraciones séricas de ácido úrico oscilaron de 0.33 mg/dL a 0.87 mg/dL.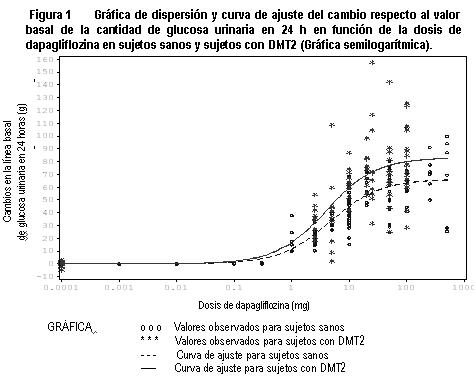
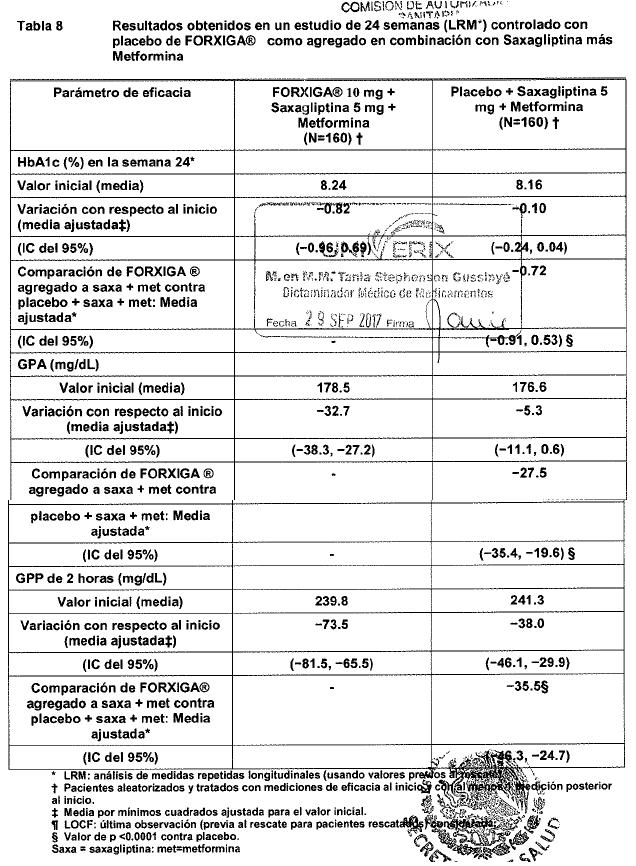
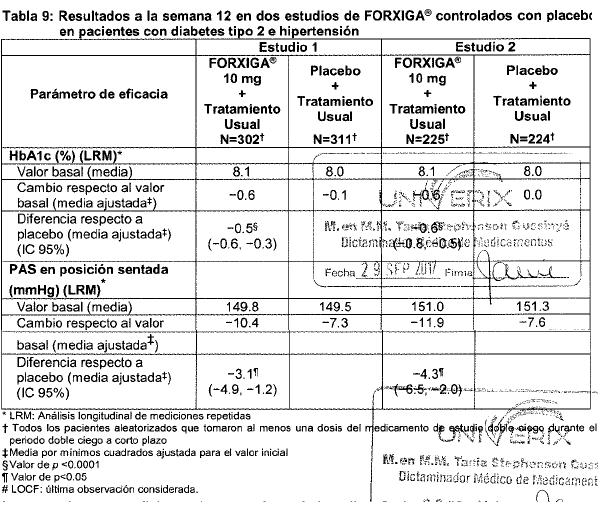
Electrofisiología cardiaca: Dapagliflozina no se asoció con la prolongación clínicamente significativa del intervalo QTc en dosis diarias de hasta 150 mg (15 veces la dosis recomendada) en un estudio de sujetos sanos. Además, no se observó ningún efecto clínicamente importante en el intervalo QTc después de dosis únicas de hasta 500 mg (50 veces la dosis recomendada) de dapagliflozina en sujetos sanos. Mecanismo de acción: Dapagliflozina es un inhibidor selectivo altamente potente y reversible del cotransportador de sodio-glucosa tipo 2 (SGLT2) que mejora el control glucémico en pacientes con diabetes mellitus tipo 2 mediante la reducción de la reabsorción de glucosa renal que conduce a la excreción urinaria de glucosa (glucosuria). FORXIGA® está disponible en forma oral y requiere de la administración una vez al día. SGLT2 se expresa en forma selectiva en el riñón sin expresión detectada en más de otros 70 tejidos incluyendo hígado, músculo esquelético, tejido adiposo, mama, vejiga y cerebro. SGLT2 es el transportador predominante responsable de la reabsorción de la glucosa proveniente del filtrado glomerular que regresa a la circulación. A pesar de la presencia de hiperglucemia en la diabetes mellitus tipo 2, la reabsorción de glucosa filtrada continúa. Dapagliflozina reduce el transporte máximo de glucosa tubular en 55% y reduce la reabsorción renal de glucosa, de tal manera que la glucosa aparece en la orina a niveles normales de glucosa plasmática. Por lo tanto, dapagliflozina mejora los niveles plasmáticos de glucosa en ayuno y postprandial reduciendo la reabsorción renal de glucosa, lo que conduce a la excreción urinaria del exceso de glucosa. Esta excreción de glucosa (efecto glucusúrico) se observa después de la primera dosis, es continua durante el intervalo de administración de 24 horas y es sostenida durante el tratamiento. La cantidad de glucosa eliminada por el riñón a través de este mecanismo depende de la concentración de glucosa en la sangre y de la tasa de filtración glomerular (GFR). De este modo, en sujetos sanos con glucosa normal, dapagliflozina tiene una baja propensión a causar hipoglucemia. Dapagliflozina no afecta a la producción normal endógena de glucosa en respuesta a la hipoglucemia. Dapagliflozina actúa en forma independiente de la secreción de insulina y de la acción de la insulina. Con el tiempo, se ha observado mejoría en la función de las células beta (HOMA-2) en estudios clínicos con dapagliflozina. La excreción urinaria de glucosa (glucosuria), inducida por dapagliflozina, está asociada con una pérdida calórica y reducción en el peso. La mayoría de la reducción en el peso fue pérdida de grasa corporal, incluyendo la grasa visceral, más que masa magra o pérdida de volumen, como se demuestra por absorciometría de rayos X de energía dual (DXA) e imágenes de resonancia magnética. La inhibición del cotransporte de glucosa y sodio por parte de dapagliflozina también se asocia con diuresis leve y natriuresis transitoria. Dapagliflozina no inhibe otros transportadores de glucosa importantes para el transporte de glucosa hacia tejidos periféricos y es 1,400 veces más selectiva para SGLT2 frente a SGLT1, el principal transportador en el intestino responsable de la absorción de glucosa. Información de estudios clínicos: FORXIGA® se ha estudiado como monoterapia y en combinación con metformina (con o sin una sulfonilurea), sulfonilurea (glimepirida), tiazolidinediona (pioglitazona), sitagliptina (con o sin metformina), saxagliptina y metformina o insulina (con o sin otro tratamiento antidiabetico oral). FORXIGA® también se ha estudiado en pacientes con diabetes tipo 2 y enfermedad cardiovascular, y en aquéllos con diabetes tipo 2 e hipertensión. Un total de 10.193 pacientes con diabetes mellitus tipo 2 fueron estudiados en 19 estudios clínicos controlados, doble ciego, para evaluar la seguridad y eficacia de FORXIGA®; en estos estudios 6,454 pacientes fueron tratados con FORXIGA®. Diceseis estudios tuvieron un periodo de tratamiento de 24 semanas de duración, dos de 12 semanas y un estudio fue de 52 semanas de duración. De los 19 estudios, 13 de ellos contaron con un periodo de extensión a largo plazo que oscilaron de 24 a 156 semanas (hasta una duración total de 208 semanas). A través de los 19 estudios clínicos, la edad media fue de 57 años (rango 18 a 92 años) y la duración media de la diabetes fue de 7 años ( < 1 a 54 años). Cincuenta y cinco por ciento (55%) de los pacientes eran hombres, de los cuales, 77% eran caucásicos, 16% asiáticos y 4% de raza negra. Setenta y siete por ciento (77%) de los pacientes tenía un IMC de ≥ 27 kg/m2. FORXIGA® también se ha estudiado en pacientes con insuficiencia renal leve (52% de la población estudiada) a moderada (12% de la población estudiada). El tratamiento con FORXIGA® como monoterapia y en combinación con metformina (con o sin una sulfonilurea), sulfonilurea (glimepirida), tiazolidinediona (pioglitazona), sitagliptina (con o sin metformina), saxagliptina y metformina o insulina (con o sin otro tratamiento antidiabetico oral), produjo mejorías clínicamente relevantes y estadísticamente significativas en el cambio medio desde el valor basal a la semana 24 en hemoglobina glucosidada (HbA1c), glucosa plasmática en ayunas (GPA) y glucosa posprandial a las 2 horas (GPP) (cuando ésta fue medida), en comparación con el control. El tratamiento con FORXIGA® iniciado de forma concomitante con saxagliptina adicionado a metformina produjo mejoras clínicamente y estadísticamente significativas en la variación media a la semana 24 con respecto al valor inicial de HbA1c en comparación con el grupo control. Estos efectos glucémicos clínicamente relevantes fueron sostenidos en los periodos de extensión a plazo de hasta 208 semanas. Se observaron reducciones en HbA1c en subgrupos, entre ellos género, edad, raza, duración de la enfermedad e IMC basal. Además en la semana 24, se observaron reducciones clínicamente relevantes y estadísticamente significativas en los cambios medios respecto al valor basal del peso corporal con los tratamientos de combinación con FORXIGA®, en comparación con el control. Las reducciones de peso corporal fueron sostenidas en extensiones de largo plazo de hasta 208 semanas. En un estudio clínico especial, la disminución en el peso se atribuyó en forma principal a la reducción en la masa grasa corporal, medida por DXA. En dos estudios con FORXIGA® 10 mg en pacientes con diabetes tipo 2 con enfermedad cardiovascular, se observaron mejorías estadísticamente significativas en HbA1c y reducciones significativas en el peso corporal y presión sanguínea sistólica en posición sentado a la semana 24 en pacientes tratados con FORXIGA® 10 mg comparados con aquéllos tratados con placebo, y se mantuvieron hasta la semana 104. En dos estudios con FORXIGA® 10 mg en pacientes con diabetes tipo 2 con hipertensión, también se observaron reducciones estadísticamente significativas en la presión sanguínea sistólica promedio en posición sentado en pacientes tratados con FORXIGA® 10 mg combinado con otro antidiabético oral y tratamiento antihipertensivo (Inhibidores de la enzima convertidora de la angiotensina (IECA) o antagonistas de los receptores de angiotensina (ARA) en un estudio e IECA o ARA más un tratamiento antihipertensivo adicional en otro estudio) comparados con aquéllos tratados con placebo a la semana 12. FORXIGA® se evaluó a 10 mg una vez al día en 17 de 19 estudios doble ciego. También se evaluaron dosis de dapagliflozina 2.5 mg y FORXIGA® 5 mg, la dosis de 2.5 mg no fue consistentemente eficaz para el control glucémico y la de 10 mg tuvo una eficacia numérica mejor, así como una seguridad similar a la de FORXIGA®5 mg. Monoterapia: Un total de 840 pacientes con diabetes tipo 2, vírgenes al tratamiento, con control inadecuado participaron en dos estudios controlados con placebo para evaluar la eficacia y seguridad de la monoterapia con FORXIGA®. En un estudio de monoterapia, un total de 558 pacientes vírgenes al tratamiento con control inadecuado de diabetes participaron en un estudio de 24 semanas con un periodo de extensión ciego controlado de 78 semanas. Después de un periodo de inducción de 2 semanas con dieta, ejercicio y placebo, 485 pacientes con HbA1c ≥ 7% a ≤ 10% se aleatorizaron a dapagliflozina 2.5 mg, FORXIGA® 5 mg o 10 mg una vez al día en la mañana (QAM, cohorte principal) o en la tarde (QPM), o únicamente placebo en la mañana. En la semana 24, el tratamiento con FORXIGA® 10 mg QAM proporcionó mejoras significativas en HbA1c y GPA en comparación con placebo (tabla 1, figura 2). En general, la administración vespertina de FORXIGA®presentó un perfil similar de seguridad y eficacia que con FORXIGA® administrada por la mañana. El cambio medio ajustado respecto al valor basal en HbA1c y la GPA fue de -0.61% y -27.0 mg/dL, respectivamente, en la semana 102 en el grupo QAM, para pacientes tratados con FORXIGA® 10 mg, y de -0.17% y -6.9 mg/dL, respectivamente, para pacientes tratados con placebo con base en los análisis de mediciones longitudinales repetidas, excluyendo los datos posteriores al rescate. La proporción de pacientes en la cohorte principal que fueron rescatados o suspendieron debido a la falta de control glucémico en la semana 24 (ajustada para el valor basal de HbA1c) fue superior con placebo (12.0%) que con FORXIGA® 10 mg (0.0%). Hacia la semana 102 (ajustada para el valor basal de HbA1c), más pacientes con placebo (44.0%) requirieron de terapia de rescate que pacientes con FORXIGA® 10 mg (35.0%).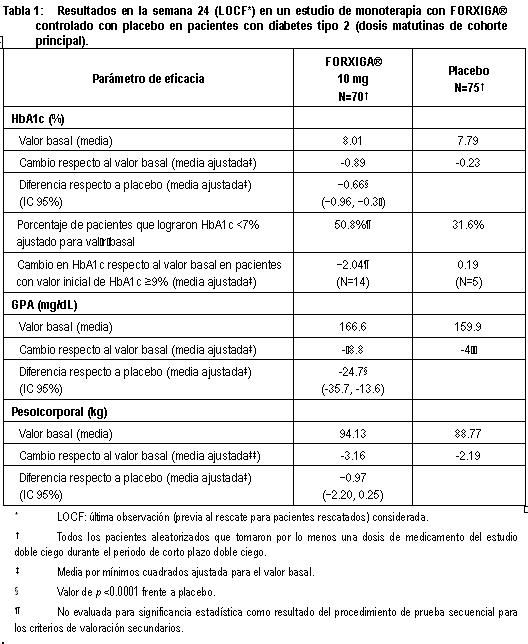
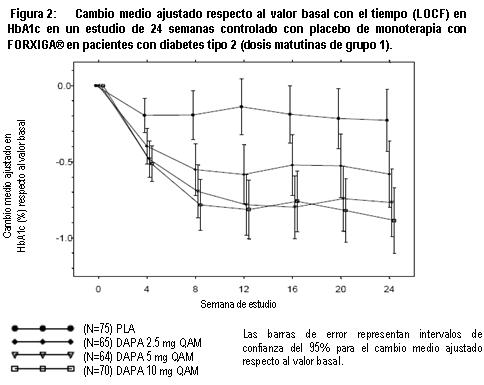
Otro estudio de 24 semanas realizado para evaluar dapagliflozina 1 mg, 2.5 mg y 5 mg en monoterapia frente a placebo, mostró mejorías clínicamente relevantes y estadísticamente significativas en los parámetros glucémicos y en el peso corporal. Terapia combinada: Se estudio FORXIGA® como tratamiento combinado inicial con metformina, y como agregado a metformina, a una sulfonilurea (glimepirida), a metformina mas una sulfonilúrea, a una tiazolidinediona (pioglitazona), a insulina (con o sin otro tratamiento antidiabetico oral), a sitagliptina (con o sin metformina), o saxagliptina mas metformina, y como tratamiento iniciado de forma concomitante con saxagliptina agregado a metformina. Terapia de combinación con metformina: Se realizaron cuatro estudios en combinación con terapia de metformina: dos estudios evaluaron a FORXIGA® adicionada a metformina como terapia de combinación inicial, un estudio evaluó el efecto de FORXIGA® adicionada a metformina en pacientes que ya tomaban metformina y en otro estudio se evaluó el efecto de FORXIGA® adicionada a metformina frente a sulfonilurea adicionada a metformina. Terapia de combinación inicial con metformina: Un total de 1,241 pacientes con diabetes tipo 2 vírgenes al tratamiento controlados en forma inadecuada (HbA1c ≥ 7.5% y ≤ 12%) participaron en dos estudios con control activo de 24 semanas de duración para evaluar la eficacia y seguridad de la terapia inicial con 5 mg o 10 mg de FORXIGA® en combinación con metformina en formulación de liberación prolongada (XR). En un estudio, 638 pacientes se aleatorizaron a uno de tres brazos de tratamiento seguidos de un periodo de inducción de 1 semana: FORXIGA® 10 mg más metformina XR (hasta 2,000 mg al día), FORXIGA® 10 mg más placebo o metformina XR (hasta 2,000 mg al día) más placebo. La dosis de metformina XR fue titulada hacia la alza semanalmente en incrementos de 500 mg, según la tolerancia, con una dosis mediana alcanzada de 2,000 mg. El tratamiento combinado de FORXIGA® 10 mg más metformina XR proporcionó mejoría significativa en HbA1c y GPA en comparación con cualquiera de los tratamientos de monoterapia y reducciones significativas en el peso corporal en comparación con metformina XR sola (tabla 2, figuras 3 y 4). FORXIGA® 10 mg como monoterapia, también brindó mejoría significativa en la GPA y reducciones significativas en el peso corporal comparado con metformina XR sola y fue no inferior a la monoterapia con metformina XR en la reducción de la HbA1c. La proporción de pacientes que fueron rescatados o que suspendieron tratamiento debido a la falta de control glucémico durante el periodo de tratamiento de 24 semanas doble ciego (ajustado para la HbA1c inicial), fue superior con el tratamiento de metformina XR más placebo (13.5%) que con FORXIGA® 10 mg más placebo y FORXIGA® 10 mg más metformina XR (7.8 y 1.4% respectivamente).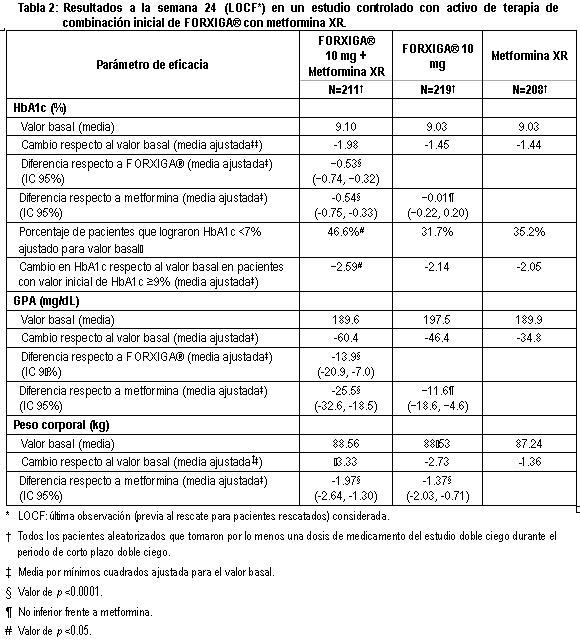
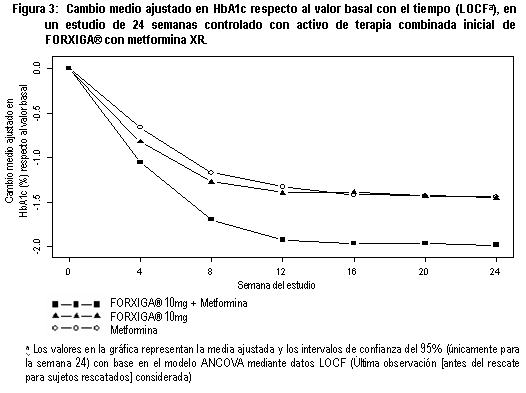
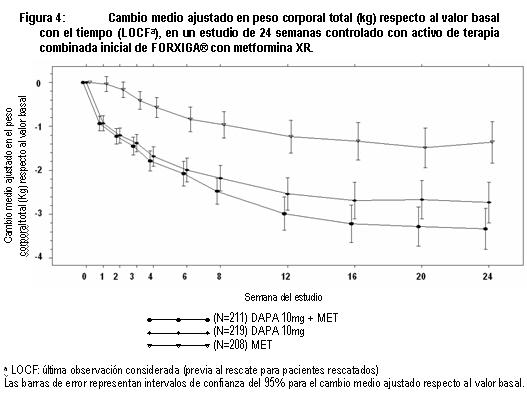
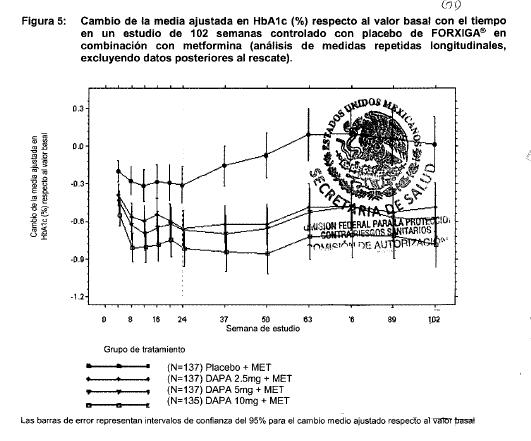
En otro estudio de 24 semanas en el que se evaluó FORXIGA® 5 mg más metformina XR, se mostraron mejorías clínicamente relevantes y estadísticamente significativas en los parámetros glucémicos frente a monoterapia de FORXIGA® 5 mg y monoterapia de metformina XR. Terapia de adición a metformina: Un total de 546 pacientes con diabetes tipo 2 con control glucémico inadecuado (HbA1c ≥ 7% y ≤ 10%) participaron en un estudio de 24 semanas controlado con placebo, con un periodo de extensión de 78 semanas, ciego controlado, para evaluar FORXIGA® adicionada a metformina. Los pacientes tratados con una dosis de por lo menos 1,500 mg al día de metformina se aleatorizaron después de completar un periodo de inducción de 2 semanas con ciego simple con placebo. Después del periodo de inducción, los pacientes aptos para su inclusión se aleatorizaron para recibir dapagliflozina 2.5 mg, FORXIGA®5 mg o 10 mg, o placebo además de su dosis actual de metformina. Como tratamiento complementario a la metformina, FORXIGA® 10 mg proporcionó mejorías significativas en HbA1c, GPA y reducciones significativa en el peso corporal en comparación con placebo en la semana 24 (tabla 3). En la semana 102, el cambio de la media ajustada respecto al valor basal en HbA1c (figura 5), GPA y peso corporal fue 0.78%, -24.5 mg/dL y -2.81 kg, respectivamente, para pacientes tratados con FORXIGA®10 mg más metformina y 0.02%, -10.4 mg/dL y -0.67 kg para pacientes tratados con placebo más metformina con base en el análisis de medidas longitudinales repetidas, excluyendo datos posteriores al rescate (figura 5). La proporción de pacientes que fueron rescatados o que suspendieron debido a la falta de control glucémico durante el periodo de tratamiento de 24 semanas doble ciego (ajustado para el valor basal de HbA1c) fue superior en el grupo de placebo más metformina (15.0%) que en el grupo de FORXIGA® 10 mg más metformina (4.4%). Hacia la semana 102 (ajustada para el valor basal de HbA1c), más pacientes tratados con placebo más metformina (60.1%) requirieron de terapia de rescate que pacientes con FORXIGA®10 mg más metformina (44.0%).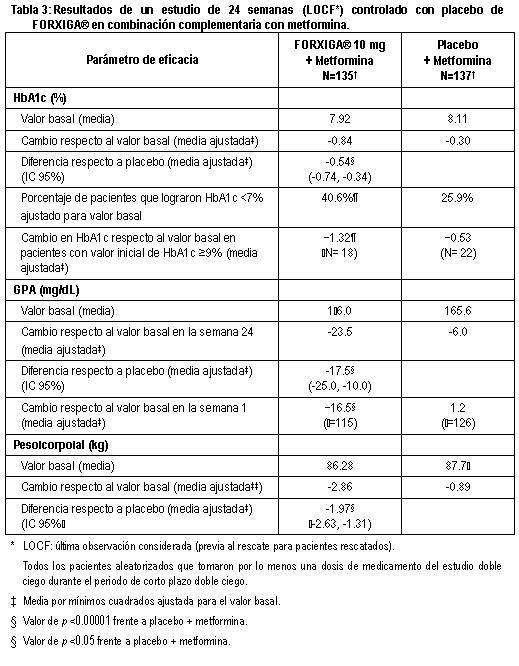
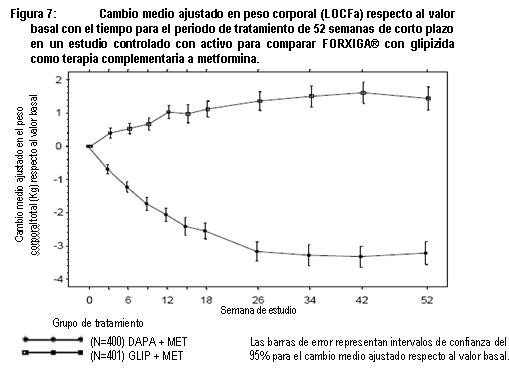
Estudio controlado con glipizida como control activo adicionada a metformina: Un total de 816 pacientes con diabetes tipo 2 con control glucémico inadecuado (HbA1c > 6.5% y ≤ 10%) se aleatorizaron en un estudio de no inferioridad de 52 semanas, controlado con glipizida, con un periodo de extensión de 156 semanas para evaluar FORXIGA® adicionada a la terapia de metformina. Los pacientes que tomaban metformina a una dosis de 1,500 mg al día se aleatorizaron después de un periodo de inducción de 2 semanas con placebo para recibir glipizida o dapagliflozina (5 mg o 2.5 mg, respectivamente) y un aumento gradual en la dosis durante 18 semanas hasta alcanzar un efecto glucémico óptimo (GPA < 110 mg/dL, < 6.1 mmol/L) o hasta el nivel de dosis más alto (hasta glipizida 20 mg y FORXIGA® 10 mg), según la tolerancia de los pacientes. Posteriormente, las dosis se mantuvieron constantes, excepto por la disminución gradual para prevenir la hipoglucemia. En este estudio no estuvo disponible el rescate por falta de control glucémico hasta la semana 104, pero lo estuvo entre las semanas 105 y 208. Al final del periodo de ajuste de la dosis, a 87% de los pacientes tratados con FORXIGA® se les ajustó la dosis hasta la dosis máxima del estudio (10 mg), frente a 73% de los tratados con glipizida (20 mg). FORXIGA®condujo a una reducción media similar en HbA1c del valor basal a la semana 52 en comparación con glipizida, con lo que se demuestra la no inferioridad (tabla 4). El tratamiento con FORXIGA® condujo a una reducción media significativa en el peso corporal del valor basal la semana 52 (LOCF) en comparación con un incremento medio en el peso corporal ocurrido en el grupo de glipizida. En la semana 104 y 208, el cambio de la media ajustada respecto al valor basal en HbA1c fue -0.32 y -0.10% y el cambio en el peso corporal fue -3.70 y -3.95 kg respectivamente para los pacientes tratados con FORXIGA® y el cambio de la media ajustada respecto al valor basal en HbA1c fue -0.14 y 0.20%, respectivamente, el cambio en el peso corporal fue 1.36 y 1.12 kg, respectivamente para pacientes tratados con glipizida, con base en el análisis de medidas longitudinales repetidas, (figuras 6 y 7). El porcentaje de pacientes que alcanzaron una pérdida de peso ≥ 5% (ajustado) en la semana 104 y 208 fue 23.8 y 10.2% respectivamente, para pacientes tratados con FORXIGA® y 2.8 y 1.8% respectivamente para pacientes tratados con glipizida. Hacia las semanas 52, 104 y 208 la proporción de pacientes que fueron suspendidos o rescatados debido a la falta de control glucémico (ajustado para el valor basal de HbA1c) fue superior con glipizida más metformina (3.6, 21.6 y 44.9% respectivamente) que con FORXIGA® más metformina (0.2, 14.5 y 39.4% respectivamente). A la semana 52, 104 y 208 respectivamente, una proporción significativamente menor de pacientes tratados con FORXIGA® (3.5, 4.3 y 5.0%) experimentaron por lo menos un evento de hipoglucemia, en comparación con glipizida (40.8, 47.0 y 50.0%).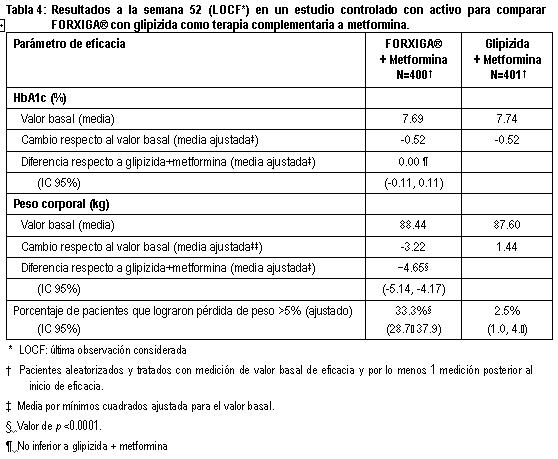

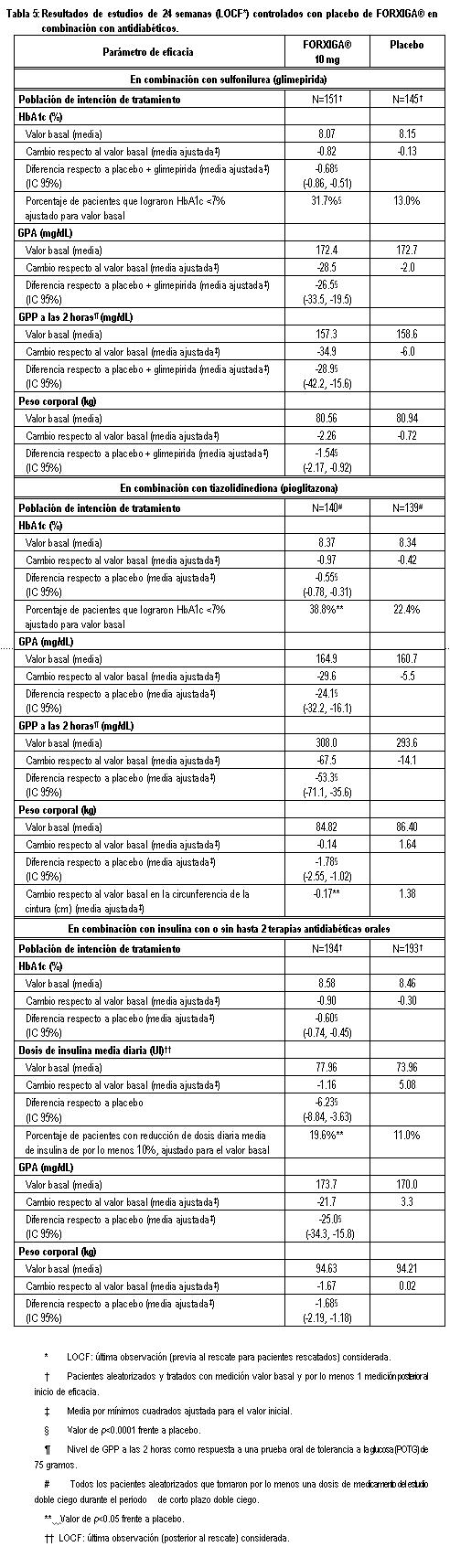
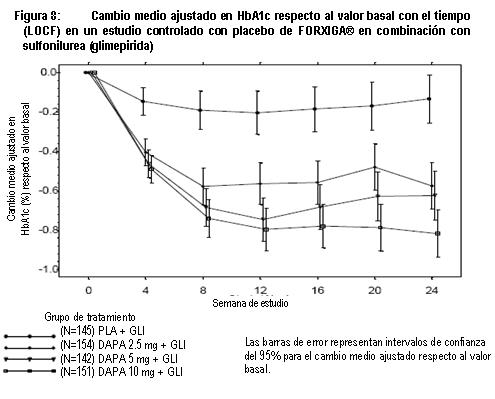
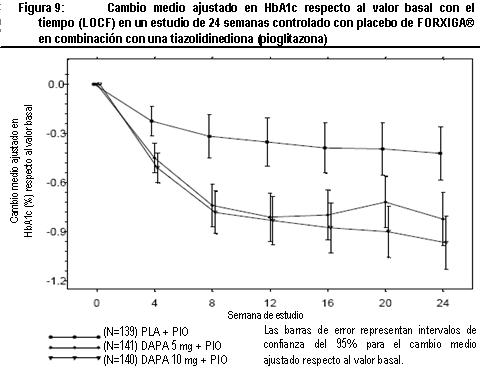
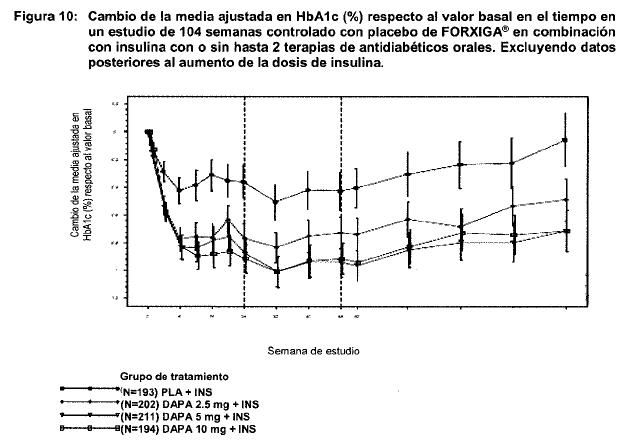
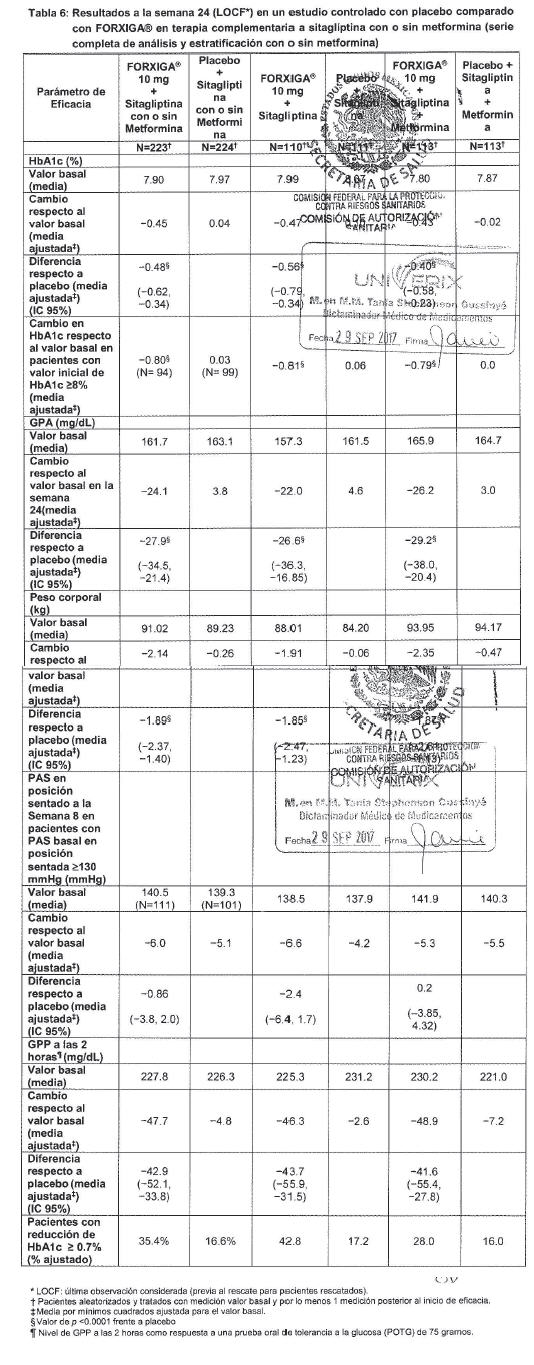
Terapia de adición con otros antidiabéticos: Terapia de adición a una sulfonilurea: Un total de 597 pacientes con diabetes tipo 2 y control glucémico inadecuado (HbA1c ≥ 7% y ≤ 10%) se aleatorizaron en este estudio de 24 semanas controlado con placebo con un periodo de extensión de 24 semanas, para evaluar FORXIGA® en combinación con glimepirida (una sulfonilurea). Los pacientes con al menos la mitad de la dosis máxima recomendada de glimepirida como monoterapia (4 mg) durante, por lo menos, 8 semanas como periodo de inducción, se aleatorizaron para recibir dapagliflozina 2.5 mg, FORXIGA® 5 mg o 10 mg o placebo adicionada a glimepirida 4 mg al día. Se permitió la disminución gradual de la dosis de glimepirida hasta 2 mg o 0 mg debido a hipoglucemia durante el periodo de tratamiento; no se permitió ajustar la dosis en aumento para glimepirida. En combinación con glimepirida, FORXIGA® 10 mg proporcionó mejoría significativa en HbA1c, GPA y GPP a las 2 horas y reducciones significativas en peso corporal en comparación con placebo más glimepirida en la semana 24 (tabla 5, figura 8). En la semana 48, el cambio de la media ajustada respecto al valor basal en HbA1c, GPA y peso corporal, fue -0.73%, -28.8 mg/dL y -2.41 kg, respectivamente, para pacientes tratados con FORXIGA® 10 mg más glimepirida y de -0.04%, 2.6 mg/dL y -0.77 kg para pacientes tratados con placebo más glimepirida en la semana 48 con base en el análisis de medidas longitudinales repetidas, excluyendo datos posteriores al rescate. A la semana 24, la proporción de pacientes que fueron rescatados o suspendidos debido a la falta de control glucémico (ajustada para el valor basal de HbA1c) fue superior con placebo más glimepirida (16.2%) que con FORXIGA® 10 mg más glimepirida (2.0%). Hacia la semana 48 (ajustada para el valor basal de HbA1c), más pacientes con placebo más glimepirida (52.1%) requirieron de terapia de rescate que pacientes con FORXIGA®10 mg más glimepirida (18.4%). Terapia de adición en combinación con metformina y una sulfonilurea: Un total de 218 pacientes con diabetes tipo 2 con control glucémico inadecuado (HbA1c ≥ 7% y ≤ 10.5%) participaron en este estudio de 24 semanas controlado con placebo con un periodo de extensión de 28 semanas, para evaluar FORXIGA® en combinación con metformina y una sulfonilurea. Los pacientes con dosis estable de metformina (formulaciones de liberación inmediata o prolongada) ≥ 1,500 mg/día más la dosis máxima tolerada, la cual debe ser al menos la mitad de la dosis máxima de una sulfonilurea, se aleatorizaron después de un periodo de inducción de 8 semanas a FORXIGA® 10 mg o placebo. No se permitió ajustar la dosis de FORXIGA® ni de metformina durante el periodo de 24 semanas de tratamiento. Se permitió el ajuste en la reducción de la dosis de sulfonilurea para prevenir hipoglucemia, pero no se permitió titular la dosis hacia arriba. En la terapia de adición en combinación con metformina y una sulfonilurea, el tratamiento con FORXIGA® 10 mg proporcionó mejorías significativas en HbA1c y GPA, así como una reducción significativa en el peso corporal en comparación con placebo en la semana 24 (tabla 5). También se observó una reducción significativa en la presión sanguínea sistólica en posición sentado en la semana 8, en pacientes tratados con FORXIGA® 10 mg en comparación con placebo. Los efectos en HbA1c, GPA y peso corporal fueron observados en la semana 52. A la semana 24, ningún paciente tratado con FORXIGA® 10 mg en combinación con metformina y una sulfonilurea y 10 pacientes (9.3%) tratados con placebo en combinación con metformina y una sulfonilurea fueron rescatados o suspendidos por falta de control glucémico (ajustado para el valor basal de HbA1c). Para la semana 52 (valor basal ajustado de HbA1c) más pacientes con placebo combinado con metformina y una sulfonilurea (44%) requirieron rescate por falta de control glucémico que pacientes con FORXIGA® (9.3%). Ningún paciente con medicamento fue retirado del estudio por falta de control glucémico. Terapia de adición a tiazolidinediona: Un total de 420 pacientes con diabetes tipo 2 con control glucémico inadecuado (HbA1c ≥ 7% y ≤ 10.5 %), participaron en este estudio de 24 semanas controlado con placebo con un periodo de extensión de 24 semanas, para evaluar FORXIGA® en combinación con pioglitazona (una tiazolidinediona) sola. Los pacientes con dosis estable de pioglitazona de 45 mg/día (o 30 mg/día, si 45 mg/día no fueron tolerados), durante 12 semanas, se aleatorizaron después de un periodo de inducción de 2 semanas a FORXIGA® 5 mg o 10 mg o placebo además de su actual dosis de pioglitazona. No se permitió ajustar la dosis de FORXIGA® ni de pioglitazona durante el estudio. En combinación con pioglitazona, el tratamiento con FORXIGA® 10 mg proporcionó mejorías significativas en HbA1c, GPP a las 2 horas y GPA, la proporción de pacientes que alcanzaron una HbA1c < 7% y una reducción significativa en el peso corporal en comparación con los grupos de tratamiento de placebo más pioglitazona (tabla 5, figura 9) en la semana 24. El tratamiento con FORXIGA® 10 mg más pioglitazona también condujo a una reducción significativa en la circunferencia de la cintura en comparación con el grupo de placebo más pioglitazona. En la semana 48, el cambio medio ajustado respecto al valor basal en HbA1c, GPA y peso corporal fue -1.21%, -33.1 mg/dL y 0.69 kg, respectivamente, para pacientes tratados con FORXIGA® 10 mg más pioglitazona y -0.54%, -13.1 mg/dL y 2.99 kg para pacientes tratados con placebo con base en el análisis de medidas longitudinales repetidas, excluyendo datos posteriores al rescate. La proporción de pacientes que fueron rescatados o se suspendieron debido a la falta de control glucémico (ajustada para el valor basal de HbA1c), fue superior en el grupo de placebo más pioglitazona (11.6%) que en el grupo de FORXIGA® 10 mg más pioglitazona (3.7%) a la semana 24. Hacia la semana 48 (ajustado para valor basal), más pacientes con placebo más pioglitazona (33.8%) requirieron de terapia de rescate que pacientes con FORXIGA® 10 mg más pioglitazona (11.8%). Terapia de adición a insulina: Un total de 808 pacientes con diabetes tipo 2 y control glucémico inadecuado (HbA1c ≥ 7.5% y ≤ 10.5%) se aleatorizaron en este estudio de 24 semanas controlado con placebo con un periodo de extensión de 80 semanas, para evaluar FORXIGA® como terapia adicionada a insulina. Los pacientes con un régimen estable de insulina, con una dosis media de al menos 30 U.I. de insulina inyectable al día, durante un periodo de por lo menos 8 semanas antes y con un máximo de dos ADO´s (antidiabético oral), entre ellos metformina, se aleatorizaron después de completar un periodo de reclutamiento de 2 semanas para recibir dapagliflozina 2.5 mg, FORXIGA® 5 mg o 10 mg o placebo, además de su actual dosis de insulina y de algún otro ADO, si aplicaba. Los pacientes se estratificaron de acuerdo con la presencia o ausencia de terapia de fondo con ADO. Se permitió el ajuste para disminuir o aumentar la dosis de insulina durante la fase de tratamiento únicamente en pacientes que no habían cumplido las metas glucémicas. No se permitieron modificaciones de dosis del medicamento ciego de estudio o del ADO durante la fase de tratamiento, con excepción de la disminución de ADO cuando hubiera preocupación acerca de la hipoglucemia después de finalizar la terapia de insulina. En este estudio, 50% de los pacientes recibieron monoterapia con insulina al inicio, mientras que el otro 50% de pacientes recibieron 1 o 2 ADO´s además de insulina. En la semana 24, FORXIGA® 10 mg proporcionó mejoría significativas en HbA1c y dosis de insulina media y reducción significativa en el peso corporal en comparación con placebo adicionada a insulina, con o sin hasta 2 ADO´s (tabla 5); el efecto de FORXIGA® en HbA1c fue similar en pacientes con insulina sola y en pacientes con insulina más ADO. En la semana 48 y 104, el cambio de media ajustada respecto al valor basal de HbA1c fue de -0.93 y -0.71%, los cambios en GPA fueron -21.5 y -18.2 mg/dL y los cambios en peso corporal -1.79 y -1.97 kg, respectivamente, para pacientes tratados con FORXIGA® 10 mg más insulin