FUZEON®
ROCHE
Denominación genérica: Enfuvirtida.
Forma farmacéutica y formulación: Solución inyectable. El frasco ámpula con liofilizado contiene: enfuvirtida 108 mg. La ampolleta con disolvente contiene: agua inyectable 2,0 ml. Los frascos ámpula de FUZEON® son monodosis, para administración subcutánea tras la reconstitución con agua esterilizada para inyectables (proporcionada en un frasco ámpula aparte). Tras la reconstitución con 1,1 ml de agua esterilizada, la solución contiene 90 mg/ml de enfuvirtida.
Indicaciones terapéuticas: FUZEON® en asociación con otros antirretrovirales está indicada para el tratamiento de la infección por VIH-1.
Farmacocinética y farmacodinamia: Farmacodinamia: mecanismo de acción: la enfuvirtida es el primer miembro del grupo terapéutico denominado inhibidores de la fusión. Se trata de un inhibidor de la reordenación estructural de la gp41 del VIH-1, que actúa uniéndose específicamente a esta proteína del virus fuera de las células y bloqueando así la entrada del virus en ellas. La enfuvirtida no precisa activación intracelular. La actividad antiviral de la enfuvirtida se debe a su asociación con la heptada repetida HR1, situada dentro de la gp41 nativa de la superficie vírica. Actividad antiviral in vitro: la actividad antiviral in vitro de la enfuvirtida ha quedado demostrada en la infección aguda de líneas celulares linfoblastoides T, células de la serie monocítica/macrofágica y células mononucleares primarias de sangre periférica (PBMC) por cepas de VIH-1 de laboratorio y clínicas. La enfuvirtida demostró actividad selectiva anti-VIH-1 frente a cepas prototípicas y primarias del virus. La sensibilidad de 130 cepas víricas basales de PBMC a la enfuvirtida se determinó en un análisis de células cMAGI de pacientes tratados con FUZEON® en los estudios clínicos de fase II. La media geométrica de la CE50 de la enfuvirtida frente a estas cepas víricas fue de 0,016g/ml (DE = 0,057), con un intervalo de valores de < 0,001 a 0,480 mg/ml. La enfuvirtida también inhibía la fusión intercelular mediada por la cubierta del VIH-1. Los estudios de la enfuvirtida en combinación con miembros representativos de los distintos grupos de antirretrovirales (inhibidores de la transcriptasa reversa, análogos nucleósidos, inhibidores de la transcriptasa reversa análogos no nucleósidos e inhibidores de la proteasa; a saber: con zidovudina, lamivudina, nelfinavir, indinavir y efavirenz) revelaron efectos aditivos o sinérgicos y la ausencia de antagonismo. No se ha establecido ninguna relación entre la sensibilidad in vitro del VIH-1 a la enfuvirtida y la inhibición de la replicación de VIH-1 en el ser humano. Dado que los blancos enzimáticos son diferentes, y como se desprende de la actividad de la enfuvirtida contra las cepas de VIH resistentes a otros grupos de antirretrovirales, las cepas de VIH resistentes a la enfuvirtida deberían permanecer sensibles a los inhibidores de la transcriptasa reversa análogos nucleósidos, los inhibidores de la transcriptasa reversa análogos no nucleósidos y los inhibidores de la proteasa. Resistencia in vitro: se han seleccionado in vitro cepas de VIH-1 con menor sensibilidad a la enfuvirtida que contienen sustituciones en los aminoácidos 36-38 del ectodominio de la gp41. Estos cambios se correlacionan con grados variables de disminución de la sensibilidad a la enfuvirtida de cepas mutantes de VIH diseñadas por mutagénesis dirigida. Resistencia in vivo: en la aparición de resistencia a la enfuvirtida influye la eficacia de todo el régimen terapéutico. Sustituciones de los aminoácidos 36-45 de gp41 durante el tratamiento se han observado en virus de pacientes tratados con FUZEON® en los estudios clínicos de fase II y III. Las sustituciones de aminoácidos observadas, en orden decreciente de frecuencia, se hallaban en las posiciones 38, 43, 36, 40, 42 y 46. No se ha establecido ninguna relación entre estas sustituciones y la eficacia in vivo del tratamiento. Los cambios en los aminoácidos 36-45 de gp41 durante el tratamiento suelen entrañar una menor sensibilidad fenotípica in vitro de las cepas víricas de estos pacientes a la enfuvirtida. Resistencia cruzada: la enfuvirtida tiene igual eficacia in vitro frente a cepas de laboratorio y clínicas en estado natural (salvajes), así como frente a las que presentan resistencia a 0, 1, 2 o 3 grupos de antirretrovirales (inhibidores de la transcriptosa reversa análogos nucleósidos, inhibidores de la transcriptosa reversa análogos no nucleósidos e inhibidores de la proteasa). Estas cepas poseían resistencia genotípica identificada específicamente frente a zidovudina, lamivudina, estavudina, didanosina, zalcitabina, abacavir, nevirapina, delavirdina, efavirenz, indinavir, saquinavir, nelfinavir, ritonavir y amprenavir; todas ellas eran sensibles a la enfuvirtida. Inversamente, las mutaciones en los aminoácidos 36-45 de gp41, que confieren resistencia a la enfuvirtida, no deberían ocasionar resistencia cruzada a otros grupos de antirretrovirales. Eficacia (estudios clínicos): estudios en pacientes tratados anteriormente con antirretrovirales: los estudios TORO-1 y TORO-2 fueron ensayos clínicos multicéntricos, aleatorizados, controlados y abiertos en sujetos infectados por el VIH-1. Los participantes tenían que presentar, bien 1) viremia a pesar de 3 a 6 meses de pretratamiento con un inhibidor de la transcriptasa reversa análogo nucleosídico (ITRAN), un inhibidor de la transcriptasa reversa análogo no nucleósido (ITRNN) o un inhibidor de la proteasa (IP), o (2) viremia y resistencia o intolerancia documentadas o por lo menos un miembro de cada uno de los grupos ITRAN, ITRNN e IP. Todos los sujetos recibieron un tratamiento de base optimizado (TO) individualizado, compuesto por 3 a 5 antirretrovirales, seleccionados de acuerdo con los tratamientos previos del sujeto y con los análisis iniciales de la resistencia vírica genotípica y fenotípica. A continuación se distribuyó aleatoriamente a los pacientes en la proporción de 2:1 para recibir 90 mg de FUZEON® 2 veces al día junto con el régimen de TO o sólo el régimen de TO. La tabla 1 resume las características demográficas de los estudios TORO-1 y TORO-2. Los sujetos habían recibido anteriormente 12 antirretrovirales (mediana) durante 7 años (mediana).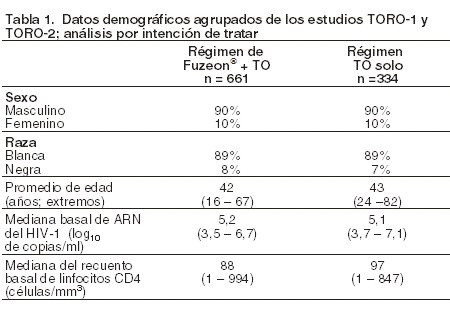
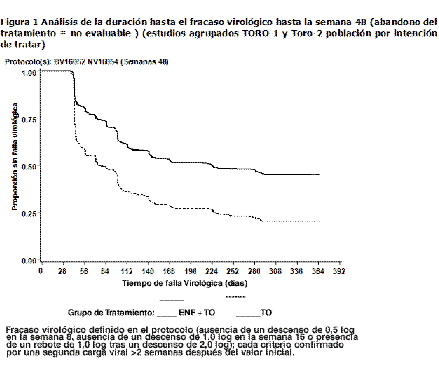
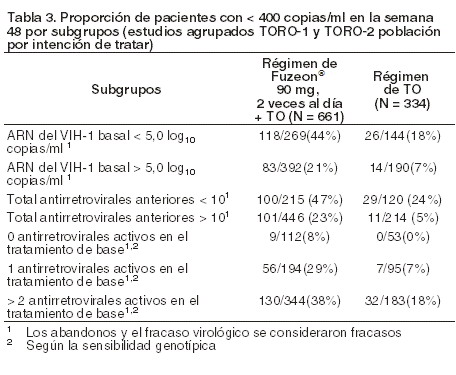
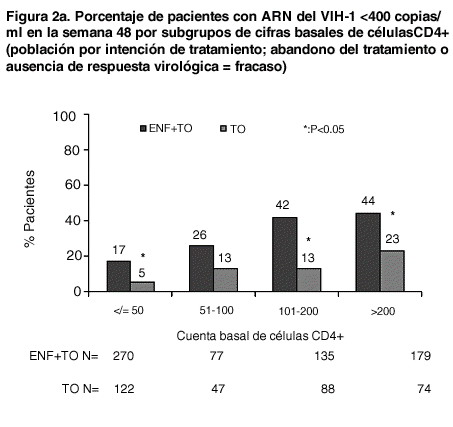
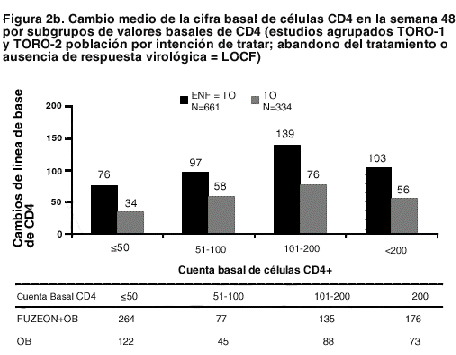
La proporción de pacientes con una carga viral < 400 copias/ml en la semana 48 fue de 30% entre los pacientes con régimen de FUZEON® + TO, frente al 12% entre los que recibían el régimen de TO solo (ver tabla 2). El tratamiento con FUZEON® + TO se asoció a una mayor proporción de pacientes que alcanzaron una cifra menor de 400 copias/ml en todos los subgrupos, considerando los valores basales de CD4 y ARN del VIH-1, el número de antirretrovirales anteriores o antirretrovirales activos en el régimen de TO. Sin embargo, en los sujetos con cifras basales de CD4 > 100 células/mm3, valores basales de ARN del VIH-1 < 5,0 log10 de copias/ml, < 10 antirretrovirales anteriores y/o 1 o más antirretrovirales en el régimen de TO era mayor la probabilidad de alcanzar una concentración de ARN del VIH-1 < 400 copias/ml con cualquiera de ambos tratamientos (ver tabla 2 y figuras 2a y 2b).
Niños: los datos disponibles sobre la eficacia de FUZEON® en niños mayores de 3 años son limitados. El estudio T20-204, en marcha, es un ensayo clínico abierto y multicéntrico para evaluar la farmacocinética, la seguridad toxicológica y la actividad antiviral de FUZEON® en 14 niños de 3 a 12 años, tratados ya con al menos 2 grupos de antirretrovirales aprobados. En el estudio T20-204, al régimen antirretroviral de base ya existente se agregó una dosis de 30 o 60 mg/m2 de FUZEON® dos veces al día. Al cabo de 7 días, el régimen de base se cambió a 3 nuevos -o sensibles- antirretrovirales y la dosis de FUZEON® se mantuvo. La mediana de edad de los pacientes era de 8 años (extremos: 3,7 y 11,9 años). La mediana basal de linfocitos CD4 era de 523/ml, la mediana basal de copias de ARN del VIH por ml era de 4,4 log10. Tras el análisis de seguridad, farmacocinética y actividad antiviral después de 7 días, todos los pacientes, salvo uno, pasaron a recibir una dosis de 60 mg/m2 de FUZEON®. La mediana de la diferencia del número de copias de ARN del VIH-1 por ml en el día 7° con respecto a la cifra basal fue de -1,15 log10 en 10 niños tratados con la dosis de 60 mg/m2. Salvo 3 pacientes, todos los demás terminaron las 48 semanas de tratamiento. En la semana 48, 6 de 14 (43%) pacientes presentaban una disminución > 1 log10 del número de copias de ARN del HIV-1 y 4 de 14 (29%) pacientes se encontraban por debajo de 400 copias/ml. En la población del análisis por tratamientos administrados, la mediana de las variaciones con respecto a los valores basales de números de copias de ARN del VIH-1 por ml y de número de linfocitos CD4 era, respectivamente, de -1,24 log10 y 237 células/ml. Farmacocinética: las propiedades farmacocinéticas de la enfuvirtida se han investigado en adultos y niños infectados por el VIH-1. Absorción: tras la inyección subcutánea en el abdomen de una dosis única de 90 mg de FUZEON® a 12 pacientes infectados por el VIH-1, la Cmáx media (± DE) era de 4,59 mg/ml ± 1,5 mg/ml; el área bajo la curva de concentraciones plasmáticas (ABC), de 55,8 mg/ml ± 12,1 mg.hr/ml, y la biodisponibilidad absoluta (utilizando la dosis IV de 90 mg como referencia), del 84,3% ± 15,5%. La absorción subcutánea de la enfuvirtida es proporcional a la dosis administrada en el intervalo de 45 a 180 mg. La absorción subcutánea de la dosis de 90 mg es comparable cuando se inyecta en el abdomen, el muslo o el brazo. La figura 3 muestra la concentración plasmática media en estado de equilibrio de la enfuvirtida en dosis de 90 mg.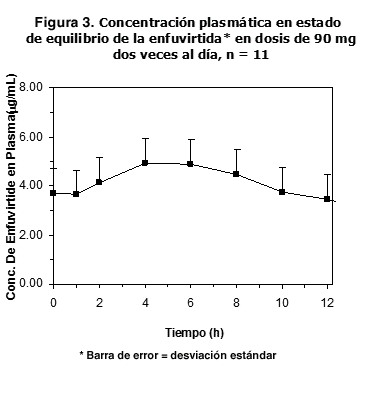
En cuatro estudios distintos (N= 9 a 12), el promedio de la concentración plasmática mínima en estado de equilibrio se situó entre 2,6 y 3,4 mg/ml. Distribución: el volumen medio (± DE) de distribución en equilibrio tras la administración intravenosa de una dosis de 90 mg de FUZEON® (N=12) era de 5,5 ± 1,1 l. La enfuvirtida se une en un 92% a las proteínas del plasma infectado por el VIH, en un intervalo de concentraciones plasmáticas de 2 a 10 mg/ml. La enfuvirtida se une sobre todo a la albúmina y, en menor medida, a la glucoproteína ácida a-1. El saquinavir, nelfinavir, lopinavir, efavirenz, nevirapina, amprenavir, itraconazol, midazolam y warfarina no desplazan a la enfuvirtida de sus sitios de unión. Por otro lado, la enfuvirtida tampoco desplaza de sus sitios de unión al efavirenz, amprenavir, midazolam ni la warfarina. Los niveles de enfurvitida en el líquido cefalorraquídeo medidos en un pequeño número de pacientes infectados con VIH-1 fueron reportados como insignificantes. La molécula puede ser muy grande para pasar la barrera hemato-encefálica. Metabolismo: la enfuvirtida -siendo un péptido- se cataboliza en sus aminoácidos constituyentes, los cuales, a continuación, se reciclan dentro del organismo. Los estudios in vitro con microsomas humanos indican que la enfuvirtida no inhibe las enzimas del citocromo P-450. En estudios in vitro con microsomas y hepatocitos humanos, la hidrólisis del grupo amida de la fenilalanina carboxiterminal da lugar a un metabolito desamidado, cuya formación no depende del NADPH. Este metabolito se detecta en el plasma humano después de administrar la enfuvirtida, con un valor de ABC que varía entre el 2,4 y el 15% del ABC de la enfuvirtida. Eliminación: no se han efectuado estudios de balance de masas para determinar la(s) vía(s) de eliminación de la enfuvirtida en el ser humano. Ahora bien, los estudios con roedores tratados con enfuvirtida radiomarcada con H3 mostraban una recuperación incompleta de la radiactividad en los excrementos en los animales al cabo de 7 días de la administración y retención de radiactividad en los músculos esqueléticos. Tras una dosis subcutánea de enfuvirtida de 90 mg (N = 12), la semivida media de eliminación (± DE) es de 3,8 ± 0,6 h, y la depuración medio (± DE), de 1,7±0,4 l/h. Farmacocinética en poblaciones especiales: insuficiencia hepática: no se ha estudiado la farmacocinética de la enfuvirtida en pacientes con insuficiencia hepática. Insuficiencia renal: el análisis de los datos de la concentración plasmática en los pacientes de los ensayos clínicos revela que la depuración de la enfuvirtida no se ve afectada en ningún grado clínico cuando la depuración de creatinina es mayor de 35 ml/min. Los resultados de un estudio de insuficiencia renal indicaron que la depuración de enfuvirtida se redujo en 38% en pacientes con disfunción renal severa y en 14-28% en pacientes con insuficiencia renal terminal mantenidos con diálisis comparada con los pacientes con función renal normal. Los resultados estuvieron dentro de los rangos vistos en los pacientes de los estudios pivote con función renal normal. La hemodiálisis no altera significativamente la depuración de enfuvirtida. Por lo tanto no se requiere ajuste de la dosis para los pacientes con disfunción renal. Sexo y peso: el análisis de los datos de la concentración plasmática en los pacientes de los ensayos clínicos puso de relieve que la depuración de la enfuvirtida es un 20% menor en el sexo femenino que en el masculino, y que aumenta con el peso corporal independientemente del sexo (20% superior en los pacientes de 100 kg y 20% inferior en los de 40kg, en relación con un paciente prototipo de 70kg). Ahora bien, esas variaciones carecen de importancia clínica y no se precisa ningún ajuste posológico. Raza: el análisis de los datos de la concentración plasmática en los pacientes de los ensayos clínicos indica que la depuración de la enfuvirtida no difiere entre las personas de raza negra y de raza blanca. Otros estudios farmacocinéticos tampoco muestran diferencias entre los asiáticos y los blancos una vez ajustada la exposición en función del peso corporal. Ancianos: no se ha estudiado la farmacocinética de la enfuvirtida en pacientes mayores de 65 años. Niños: se ha estudiado la farmacocinética de la enfuvirtida en 32 niños y adolescentes de 3 a 16 años, tratados con dosis de 0,5 a 2,5 mg/kg. La dosis de 2 mg/kg dos veces al día (máximo de 90 mg dos veces al día) produjo concentraciones plasmáticas de enfuvirtida similares a las obtenidas en adultos tratados con 90 mg dos veces al día. En 25 niños y adolescentes de 5 a 16 años tratados con una dosis de 2 mg/kg dos veces al día, se obtuvieron los valores siguientes: ABC medio en equilibrio estable, 54,3 ± 23,5 mg.h/ml; Cmáx 6,14 ± 2,48 mg/ml, y Cmín 2,93 ± 1,55 mg/ml.
Contraindicaciones: FUZEON® está contraindicado en las personas alérgicas a la enfuvirtida o a cualquier otro de sus componentes.
Precauciones generales: Como los demás antirretrovirales, FUZEON® debe administrarse como parte de un régimen combinado. El tratamiento con FUZEON® se ha asociado ocasionalmente con reacciones de hipersensibilidad; en raras ocasiones, estas reacciones han recidivado tras la reexposición. Los acontecimientos adversos consistían en exantema, fiebre, náuseas o vómitos, escalofríos, hipotensión y elevación de las transaminasas con algunas combinaciones medicamentosas, así como posiblemente reacción primaria mediada por inmunocomplejos, dificultad respiratoria y gromerulonefritis. Los pacientes con signos o síntomas de hipersensibilidad sistémica deberán suspender la administración de FUZEON® y acudir inmediatamente al médico para una evaluación clínica. El tratamiento con FUZEON® no debe reinstaurarse si los signos o síntomas sistémicos son los de una reacción de hipersensibilidad que se considere relacionada con FUZEON®. No se conocen factores de riesgo anticipatorios del desarrollo de hipersensibilidad a FUZEON®. Entre los pacientes tratados con FUZEON® en los estudios clínicos se ha observado un aumento de la incidencia de neumonía bacteriana, letal en algunos casos. Los factores de riesgo de neumonía fueron los siguientes: recuento basal de linfocitos CD4 bajo, carga viral basal alta, uso de drogas por vía intravenosa, tabaquismo y antecedentes de enfermedad pulmonar. Se vigilará estrechamente a los pacientes para detectar si se presentan signos o síntomas de infección, sobre todo si sufren enfermedades subyacentes que puedan predisponer al desarrollo de una neumonía. La administración de FUZEON® a personas no infectadas por el VIH-1 (por ejemplo profilaxis tras la exposición) puede inducir la formación de anticuerpos contra la enfuvirtida que desencadenen una reacción cruzada con la gp 41 del VIH. Esto podría ocasionar un resultado falso positivo de la prueba ELISA de anticuerpos anti-VIH. Síndrome de reconstitución inmune (también referido como síndrome de reactivación inmune, enfermedad de restauración inmune o síndrome de inflamación y reconstitución inmune): se ha reportado al síndrome de reconstitución inmune en pacientes tratados con terapia antirretroviral de combinación, incluyendo a FUZEON®. Durante la fase inicial del tratamiento de combinación antirretroviral, los pacientes cuyo sistema inmune responde, pueden desarrollar una respuesta inflamatoria a infecciones oportunistas residuales o inactivas (como la infección a Mycobacterium avium, citomegalovirus, neumonía por Pneumocystis jirovecii [NPC], tuberculosis u otras), que puedan necesitar evaluación inmediata y tratamiento. Efectos sobre la capacidad para conducir y utilizar máquinas: no se han realizado estudios sobre la capacidad para conducir vehículos y utilizar las máquinas durante el tratamiento con FUZEON®. Aunque no hay indicios de que FUZEON® pueda alterar la capacidad del paciente para conducir vehículos o manejar máquinas, deben tenerse en cuenta los acontecimientos adversos de FUZEON® (ver Reacciones secundarias y adversas).
Restricciones de uso durante el embarazo y la lactancia: La enfuvirtida no causó reacciones adversas en el desarrollo embrionario en los estudios de teratogenia realizados en ratas y conejos, animales a los que se expuso a dosis 8,9 veces mayores que las dosis terapéuticas previstas para el ser humano. Ahora bien, no se han efectuado estudios adecuados y bien controlados en mujeres embarazadas. Por consiguiente, sólo se debe utilizar FUZEON® durante el embarazo cuando los beneficios esperados justifiquen el riesgo potencial. Después de administrar enfuvirtida radiomarcada con H3 a ratas lactantes, se detectó en la leche una cantidad muy baja de radiactividad. Se ignora si la enfuvirtida pasa a la leche materna humana. Por ello, conviene advertir a las madres que no amamanten a sus hijos si están recibiendo FUZEON®, ante la posibilidad de transmisión del VIH y de que el niño lactante sufra reacciones adversas.
Reacciones secundarias y adversas: Experiencia adquirida en los estudios clínicos: las características generales de seguridad toxicológica de FUZEON® se basan en los datos de 2.120 pacientes que recibieron como mínimo una dosis de enfuvirtida durante diversos ensayos clínicos. La población de estudio estaba integrada por 2.051 adultos (incluidos 1.181 que habían recibido la dosis recomendada durante ≥24 semanas y 631 que la habían recibido durante ≥48 semanas) y 69 niños (incluidos 44 que habían recibido FUZEON® durante ≥24 semanas y 27 que lo habían recibido durante ≥48 semanas). Adultos: el análisis primario de seguridad toxicológica en adultos se basa en los resultados conjuntos después de 48 semanas de dos estudios controlados y aleatorizados de fase III (TORO-1 y TORO-2) en adultos infectados por el VIH-1 pretratados y/o con resistencia documentada previa y/o con intolerancia a los inhibidores de la proteasa, los inhibidores de la transcriptasa reversa análogos nucleósidos o los inhibidores de la transcriptasa reversa análogos no nucleósidos. Los pacientes fueron distribuidos aleatoriamente en la proporción 2:1 para recibir FUZEON® por vía subcutánea en una dosis de 90 mg dos veces al día en asociación con un tratamiento antirretroviral de base optimizado (TO) (n=663 pacientes) o, como grupo de control, solamente los antirretrovirales del tratamiento de base optimizado (n=334). En los estudios TORO-1 y TORO-2, al finalizar la semana 8 se permitió que, en los pacientes con el régimen de TO solo que cumplían los criterios definidos en el protocolo, se revisara el tratamiento de base y se agregara FUZEON®. En la semana 48 de estudio, la exposición acumulativa con el régimen de FUZEON® + TO era de 557 años-paciente, y con el régimen TO solo, de 162 años-paciente. Debido a esta diferencia de exposición, los resultados de seguridad ajustados se expresan como número de pacientes con un determinado acontecimiento adverso por 100 años-paciente de exposición (excepto para las reacciones en la zona de inyección). Reacciones en el sitio de inyección: las reacciones secundarias más frecuentes tras la administración de FUZEON® consistieron en reacciones en el sitio de la inyección (RSI), registradas en un 98% de los 663 pacientes de TORO-1 y TORO-2 (tabla 4). Tan sólo el 4% de los pacientes dejaron de recibir FUZEON® a causa de RSI. La inmensa mayoría (85% de TORO-1 y TORO-2) de las RSI se produjeron en la primera semana de administración de FUZEON® y estaban asociados con dolor o molestias de carácter leve o moderado en la zona de inyección, pero sin obligar a los pacientes a restringir las actividades habituales. La intensidad del dolor o las molestias asociadas con las RSI no aumentó en el curso del tratamiento. Los signos y síntomas característicos de las RSI tuvieron una duración, por lo general ≤7 días y el número de lesiones detectadas en cualquiera de las visitas del estudio fue ≤5 en el 72% de los pacientes con lesiones detectadas. Las infecciones en el lugar de inyección (incluidos los abscesos y la celulitis) afectaron al 1,5% de los pacientes.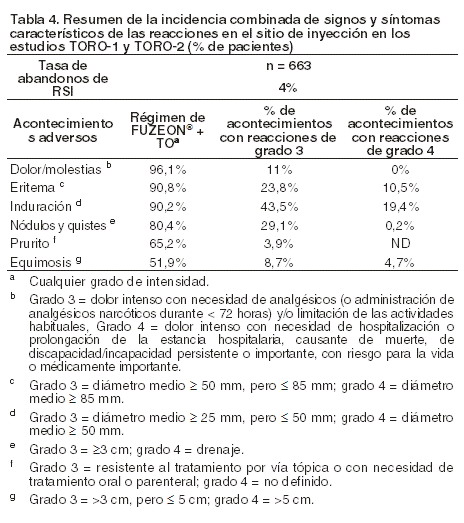
Otras reacciones adversas: los acontecimientos adversos notificados con mayor frecuencia en el grupo tratado de FUZEON® + TO (n=663), excluidas las reacciones en el sitio de la inyección, fueron diarrea (38 pacientes por 100 años-paciente) y náuseas (27 pacientes por 100 años-paciente). Estos acontecimientos adversos también se observaron habitualmente en los que recibieron el régimen de TO solo (n=334): diarrea (73 pacientes por 100 años-paciente) y náuseas (50 pacientes por 100 años-paciente). La adición de FUZEON® al tratamiento antirretroviral de base no aumentó, en general, la frecuencia ni la gravedad de la mayoría de los acontecimientos adversos. La tabla 5 recoge los acontecimientos registrados con más frecuencia con el régimen de FUZEON® + TO (n=663) que en el régimen de TO solo (n=334) (excluidas las reacciones en el sitio de inyección), con una tasa ajustada por la exposición de al menos 2 pacientes por 100 años-paciente (datos de los estudios clínicos TORO-1 y TORO-2). Las tasas de acontecimientos adversos en los pacientes que cambiaron a FUZEON®, tras un fracaso terapéutico, fueron similares. Los dos únicos acontecimientos adversos con una razón de riesgos estadísticamente significativa entre el régimen con FUZEON® y el régimen de TO solo fueron neumonía y linfadenopatía. La mayoría de los acontecimientos adversos fueron de intensidad leve o moderada.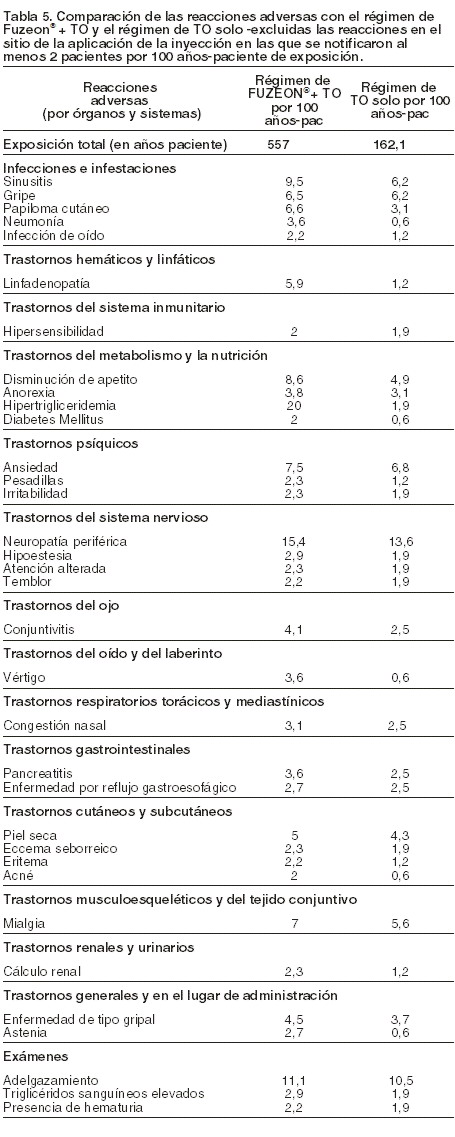
Se han registrado, además, un pequeño número de reacciones de hipersensibilidad atribuidas a FUZEON® las cuales en raras ocasiones han recidivado tras la reexposición (ver Precauciones generales). Se registró una tasa más alta de neumonía bacteriana (se incluyó en el análisis la bronconeumonía y acontecimientos relacionados) entre los tratados con el régimen de FUZEON® + TO en los estudios TORO-1 y TORO-2 que en el grupo de control con el régimen de TO solo (6,6 pacientes con episodios de neumonía por 100 años-pacientes vs. 0,6 pacientes con episodios de neumonía por 100 años-pacientes, respectivamente). La incidencia observada entre los pacientes tratados con enfuvirtida concordaba con la tasa indicada en la bibliografía para esta población de pacientes. La incidencia en el grupo de control fue menor que la tasa señalada en la bibliografía. Los factores de riesgo de neumonía fueron los siguientes: recuento basal de linfocitos CD4 bajo, carga viral basal alta, uso de drogas por vía intravenosa, tabaquismo y antecedentes de enfermedad pulmonar. No está claro si la tasa más alta de neumonía estaba relacionada con FUZEON®. Se vigilará estrechamente a los pacientes para detectar si se presentan signos o síntomas de infección, sobre todo si sufren enfermedades subyacentes que puedan predisponer al desarrollo de una neumonía (ver Precauciones generales). Alteraciones de laboratorio: la mayoría de los pacientes no experimentaron cambios en el grado de toxicidad de ninguno de los parámetros de laboratorio a lo largo del estudio. La tabla 6 muestra las alteraciones de laboratorio registradas durante el tratamiento que afectaron como mínimo a 2 pacientes por 100 años-pacientes de exposición y que se produjeron con mayor frecuencia (como anomalía analítica de grado 3 o 4) en los tratados con FUZEON® + TO que en los que recibieron TO únicamente (datos agrupados de los estudios TORO-1 y TORO-2 de 48 semanas). La eosinofilia registrada hasta la semana 48 de tratamiento [recuento de eosinófilos superior al límite superior de la normalidad (LSN) de ≥0,7 x 109/l] afectó a una tasa mayor de pacientes del grupo FUZEON® + TO (12,9 por 100 años-paciente) que del régimen de TO solo (5,6 por 100 años-paciente). Si se aplica un umbral de eosinofilia más alto ( > 1,4 x 109/l), la tasa ajustada por la exposición es similar en ambos grupos (2,2 y 1,8 pacientes con eosinofilia por 100 años-paciente con los regímenes de FUZEON® + TO y TO solo, respectivamente).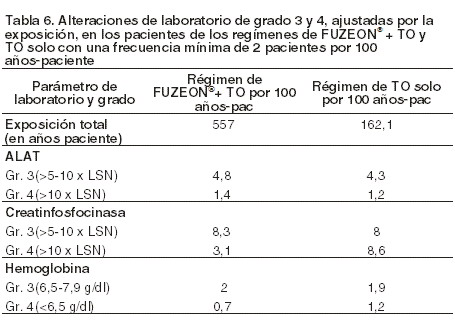
Otros acontecimientos adversos u otras alteraciones analíticas: los acontecimientos adversos o las alteraciones de laboratorio siguientes se notificaron asimismo en el análisis de 24 semanas de los dos estudios fundamentales con una incidencia > 2% y una frecuencia mayor con los pacientes tratados con FUZEON® + TO que en los que habían recibido el régimen de TO solo. No se ha establecido una relación causal entre estos acontecimientos adversos y FUZEON®. Infecciones e infestaciones: candidiasis oral, herpes simple, foliculitis. Trastornos psíquicos: insomnio, depresión. Trastornos del sistema nervioso: cefalea, mareo (excluyendo vértigo), disgeusia. Trastornos respiratorios, torácicos y mediastínicos: tos. Trastornos gastrointestinales: dolor en la zona superior del abdomen, estreñimiento, dolor faríngeo. Trastornos cutáneos y subcutáneos: prurito, sudores nocturnos, sudoración aumentada. Trastornos musculoesqueléticos y del tejido conjuntivo: artralgia, dorsalgia, dolor en las extremidades y calambres musculares. Trastornos generales y alteraciones en el sitio de la administración: astenia. Exámenes: valores elevados de la gamma-glutamiltransferasa, amilasa, lipasa y AST. Niños: se ha estudiado FUZEON® en 69 niños de 4 a 16 años, con una exposición al medicamento entre 1 dosis y > 48 semanas de tratamiento. Los acontecimientos adversos registrados durante los ensayos clínicos eran similares a los observados en sujetos adultos.
Interacciones medicamentosas y de otro género: FUZEON® no se debe mezclar con otros medicamentos, salvo con el disolvente incluido en el envase (agua inyectable). No se conocen interacciones farmacocinéticas clínicamente relevantes entre la enfuvirtida y otros medicamentos administrados a la vez y metabolizados por las enzimas del citocromo P-450. Efectos de la enfuvirtida en el metabolismo de otros fármacos: según los resultados de un estudio in vitro con microsomas humanos, la enfuvirtida no inhibe las enzimas del citocromo P-450 y, en consecuencia, no altera el metabolismo de los medicamentos metabolizados por el sistema enzimático CYP450. En un estudio in vivo del metabolismo humano, FUZEON® en la dosis recomendada de 90 mg dos veces al día no inhibió el metabolismo de los sustratos por las isoenzimas CYP3A4 (dapsona), CYP2D6 (debrisoquina), CYP1A2 (mefenitoína), CYP2C19 (mefenitoína) y CYP2E1 (clorzoxazona). Efectos de fármacos administrados simultáneamente en el metabolismo de la enfuvirtida: en estudios independientes de interacciones farmacocinéticas, la coadministración de ritonavir y rifampicina no produjo interacciones farmacocinéticas de interés clínico con FUZEON® (ver tabla 7).
Alteraciones en los resultados de pruebas de laboratorio: La mayoría de los pacientes no experimentaron cambios en el grado de toxicidad de ninguno de los parámetros de laboratorio a lo largo del estudio. La tabla 8 muestra las alteraciones analíticas registradas durante el tratamiento que afectaron como mínimo a un 2% de los pacientes y que fueron más frecuentes en aquellos tratados con FUZEON® + TO (ya sea alteraciones de laboratorio Grado 3 o 4) que en los que recibieron TO únicamente a la semana 48 de los datos agrupados de los estudios TORO-1 y TORO-2. La eosinofilia surgida durante el tratamiento hacia la semana 48 [recuento de eosinófilos mayor al límite superior normal (LSN) de ≥0,7 y 109/l] afectó a un porcentaje mayor de los pacientes tratados con enfuvirtida+TO (12,9%) que de los que recibieron TO (5,6%). Cuando se usó un umbral más alto para eosinofilia ( < 1,4 * 109/l), la frecuencia ajustada de eosinofilia es similar en ambos grupos (2,2 y 1,8% de pacientes en brazo de enfuvirtida + TO y en el esquema de TO respectivamente).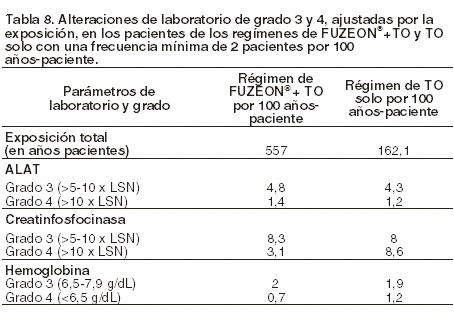
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Carcinogénesis: no se han realizado estudios de carcinogenicidad a largo plazo de la enfuvirtida. Mutagénesis: la enfuvirtida no fue mutágena ni clastógena en una serie de ensayos efectuados in vivo e in vitro, a saber: prueba de mutagenicidad de Ames, ensayo de mutación genética con células AS52 de ovario de hámster chino y prueba de micronúcleos en el ratón in vivo. Trastornos de la fecundidad: la enfuvirtida no tuvo efectos adversos sobre la fertilidad de ratas macho y hembra en dosis 0,7, 2,5 y 8,3 veces superiores a la máxima diaria recomendada para el ser humano (en mg/kg), administrada en inyección subcutánea. Teratogénesis: la enfuvirtida no causó reacciones adversas en el desarrollo embrionario en los estudios de teratogenia realizados en ratas y conejos, a los que se expuso a dosis de 8,9 veces mayores que las dosis terapéuticas previstas para el ser humano. Reacciones locales en el lugar de inyección: en macacos de Java (monos Cynomolgus) tratados con enfuvirtida en dosis de 5 y 10 mg/kg dos veces al día durante nueve meses, las reacciones en la zona de inyección consistieron en tumefacción, edema y abscesos observadas generalmente después del quinto mes. En el examen necroscópico, los efectos más prominentes en los sitios de inyección de la enfuvirtida fueron decoloración, engrosamiento y quistes. Microscópicamente se detectaron hemorragia subcutánea, edema e infiltrado inflamatorio, presentes en proporción e intensidad marcadamente mayor en los sitios de inyección de la enfuvirtida. El infiltrado inflamatorio mixto era predominantemente linfocítico y formaba con frecuencia folículos linfoides. La proporción de células plasmáticas era sustancial, lo cual concuerda con la observación de una producción crónica persistente de anticuerpos contra la enfuvirtida, y se observaron también en el infiltrado polimorfonucleares eosinófilos, que sugerían una reacción de hipersensibilidad. En cerdos miniatura tratados hasta dos semanas con enfuvirtida (4 inyecciones/día de 50 mg/ml o 2 inyecciones/día de 100 mg/ml) se detectaron masas subcutáneas en el espacio de 8 días de inyecciones diarias repetidas. Los exámenes microscópicos de biopsias en sacabocado de los sitios de inyección los días 8 y 15 revelaron edema, inflamación aguda o crónica, necrosis, fibrosis y degeneración colágena en la dermis. Se observaron cambios prominentes en la dermis y el tejido subcutáneo, que incluían la presencia de células gigantes multinucleadas.
Dosis y vía de administración: Dosis y forma de administración: los pacientes y los profesionales de la salud que apliquen FUZEON® deben estar instruidos en el uso de técnicas asépticas cuando requieran administrar FUZEON® para evitar infecciones en el sitio de inyección (ver Reacciones secundarias y adversas). Se deben proporcionar las instrucciones apropiadas para la reconstitución de FUZEON® y su autoaplicación. Se recomienda que la primera inyección se realice bajo supervisión de un profesional de la salud calificado. Se recomienda que los pacientes comprendan el uso de técnicas asépticas de autoaplicación y que los procedimientos se reevalúen periódicamente. Los pacientes y los profesionales de la salud deben estar instruidos con las técnicas apropiadas de preparación de la inyección y del desecho de las jeringas y agujas para evitar accidentes. Los pacientes deben estar advertidos respecto a la reutilización de las jeringas y agujas, instruidos en los procedimientos seguros para desechar los materiales incluyendo el uso de un contenedor de materiales punzo-cortantes desechables. Los pacientes deben instruirse respecto al desecho de estos contenedores. En caso de ocurrir algún accidente con las agujas utilizadas por un paciente, acudir de inmediato al médico. FUZEON® no contiene conservadores. FUZEON® se presenta en la forma de polvo liofilizado, el cual ha de reconstituirse con agua esterilizada e inyectarse por vía subcutánea. Reconstitución y uso: FUZEON® debe ser reconstituido solamente con 1,1 ml de agua estéril para inyección. Después de agregar el agua estéril, debe ser golpeado ligeramente por 10 segundos rodado entre las manos para evitar formación de espuma y asegurar que todas las partículas del medicamento entren en contacto con el líquido y no queden restos de medicamento en las paredes del vial. El vial debe ponerse en reposo hasta que el polvo se disuelva completamente, lo cual puede tardar hasta 45 minutos. El tiempo de reconstitución puede reducirse rodando el vial suavemente entre las manos hasta total disolución. Antes de administrar el producto se debe inspeccionar visualmente el vial para asegurar que el contenido está disuelto, que la solución sea clara, incolora, sin burbujas o materia particulada. Si hay evidencia de materia particulada, no debe usarse el vial y debe devolverse a la farmacia. Una vez reconstituido FUZEON® debe administrarse inmediatamente o, si la solución de FUZEON® no puede inyectarse inmediatamente, se debe mantener en refrigeración en el vial original hasta el momento de usar, y utilizar en el espacio de 24 horas. La dosis subsiguiente de FUZEON® debe ser reconstituida por adelantado y almacenarse en el refrigerador en el vial original hasta el momento de usar y utilizar en el espacio de 24 horas. La solución reconstituida refrigerada debe llevarse a temperatura ambiente antes de la inyección y el vial debe ser inspeccionado visualmente otra vez para asegurar que los contenidos están completamente disueltos y que la solución sea clara, incolora, sin burbujas o materia particulada. La solución reconstituida debe inyectarse subcutáneamente en el brazo proximal, abdomen o muslo anterior. La inyección no debe aplicarse en el mismo sitio de inyección o donde cause reacción. Asimismo no inyectar en lunares, cicatrices, contusiones o en el ombligo. Dosis habitual: adultos: la dosis recomendada de FUZEON® es de 90 mg dos veces al día en inyección subcutánea en el brazo, la cara anterior del muslo o el abdomen. El lugar de la inyección debe ser diferente al de la inyección precedente y no presentar reacciones locales en la zona de administración. Pautas posológicas especiales: niños: no hay datos suficientes para establecer recomendaciones posológicas de FUZEON® en niños menores de 6 años. Para niños y adolescentes de 6 a 16 años de edad, la dosis recomendada se sitúa entre 2 mg/kg dos veces al día y un máximo de 90 mg dos veces al día, administrada en inyección subcutánea en el brazo, la cara anterior del muslo o el abdomen (ver tabla 9). El lugar de la inyección debe ser diferente al de la inyección precedente y no presentar reacciones locales en la zona de administración.
Uso geriátrico: los estudios clínicos de FUZEON® no incluyeron un número suficiente de pacientes mayores de 65 años de edad para determinar si responden de manera diferente a pacientes más jóvenes. Insuficiencia renal: no se recomienda ajustar la dosis en los pacientes con insuficiencia renal, incluyendo aquellos que reciben hemodiálisis. Insuficiencia hepática: no hay datos disponibles para establecer recomendaciones posológicas en pacientes con insuficiencia hepática.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: No se han descrito casos de sobredosis de FUZEON® en el ser humano. La dosis máxima administrada a 12 pacientes en un ensayo clínico (estudio T20-501) fue de 180 mg en inyección subcutánea única. Estos pacientes no experimentaron ningún acontecimiento adverso que no se haya observado en la dosis recomendada. En un estudio del Programa de acceso temprano, un paciente recibió en una ocasión 180 mg de FUZEON® en una dosis única, pero no experimentó por ello ningún efecto adverso. No se conoce ningún antídoto específico para FUZEON®. En caso de sobredosis, se aplicarán las medidas de apoyo habituales.
Presentación(es): Caja con 60 frascos ámpula con liofilizado, 60 frascos ámpula