GALVUS
NOVARTIS
Denominación genérica: Vildagliptina
Forma farmacéutica y formulación: Cada comprimido contiene: Vildagliptina 50 mg Excipiente cbp 1 comprimido
Indicaciones terapéuticas: GALVUS® está indicado como complemento de la dieta y el ejercicio para mejorar el control glucémico en pacientes con diabetes mellitus tipo 2. En monoterapia. En terapia combinada. Con metformina, cuando la dieta, el ejercicio y la monoterapia con metformina no permitan conseguir un control adecuado de la glucemia. Con una sulfonilurea (SU), cuando la dieta, el ejercicio y la monoterapia con la SU no permitan conseguir un control adecuado de la glucemia. Con una tiazolidinediona (TZD), cuando la dieta, el ejercicio y la monoterapia con la TZD no permitan conseguir un control adecuado de la glucemia. En terapia triple. Con una sulfonilurea y metformina, cuando la dieta y el ejercicio más la biterapia dual con estos fármacos no permitan conseguir un control adecuado de la glucemia. GALVUS® también está indicado en terapia combinada con insulina (con o sin metformina), cuando la dieta, el ejercicio y una dosis estable de insulina no permitan conseguir un control adecuado de la glucemia. GALVUS® está indicado también como terapia de combinación inicial con Metformina en pacientes con Diabetes Mellitus Tipo 2 cuando la diabetes no esté adecuadamente controlada sólo por dieta y ejercicio.
Farmacocinética y farmacodinamia: Grupo farmacoterapéutico, código ATC: lnhibidor de la dipeptidil peptidasa 4 (DPP-4) para el tratamiento de la diabetes. Código ATC: A10BH02. Mecanismo de acción: La vildagliptina pertenece a la clase de los potenciadores de los islotes pancreáticos y mejora el control de la glucemia mediante la inhibición potente y selectiva de la dipeptidil-peptidasa 4 (DPP-4). Tal inhibición aumenta las concentraciones endógenas en ayunas y posprandiales de las hormonas incretinas GLP-1 (péptido 1 similar al glucagón) y GIP (polipéptido insulinotrópico dependiente de la glucosa). Farmacocinética: Absorción: Tras la administración oral en ayunas del medicamento, la vildagliptina se absorbe rápidamente y se observan concentraciones plasmáticas máximas a las 1.75 horas. La administración concomitante con alimentos reduce levemente la velocidad de absorción de vildagliptina, lo cual se caracteriza por una disminución de la concentración máxima en un 19% y retraso en el tiempo a concentración plasmática máxima a 2.5 horas. No existe cambio en la proporción de absorción, los alimentos no afectan la exposición general (ABC). Distribución: La unión de vildagliptina a proteínas plasmáticas es baja (9.3%); la vildagliptina es distribuida uniformemente entre el plasma y los eritrocitos. El volumen medio de distribución de vildagliptina en el estado estable tras la administración intravenosa (VSS) es 71 Litros, lo cual es un indicio de distribución extravascular. Biotransformación/metabolismo: En los seres humanos, vildagliptina se elimina principalmente por vía metabólica (69% de la dosis). El metabolito principal, LAY151, es farmacológicamente inactivo, se forma por hidrólisis del grupo ciano y da cuenta del 57% de la dosis: le sigue en importancia el producto de la hidrólisis amídica, que representa el 4% de la dosis. Un estudio realizado en ratas deficientes con respecto a DPP4, in vivo, ha revelado que la DPP4 participa parcialmente en la hidrólisis de vildagliptina. Las enzimas del citocromo P450 no metabolizan la vildagliptina en grado cuantificable alguno. Los estudios in vitro indican que la vildagliptina no inhibe ni induce las enzimas del citocromo P450. Eliminación: Tras la administración oral de [14C]-vildagliptina, cerca del 85% de la dosis se excreta en la orina y el 15% de la dosis se recupera en las heces; la excreción renal de vildagliptina inalterada representa el 23% de la dosis. Tras la administración intravenosa a individuos sanos, la depuración renal y plasmática total de vildagliptina es de 41 Litros/hora y 13 Litros/hora, respectivamente. La vida media de eliminación promedio tras la administración intravenosa es aproximadamente igual a 2 horas. La vida media de eliminación tras la administración oral es de unas 3 horas e independiente de la dosis. Linealidad: La vildagliptina se absorbe rápidamente con una biodisponibilidad oral absoluta de 85%. Las concentraciones plasmáticas máximas de vildagliptina y el área bajo la curva de concentraciones plasmáticas sobre el tiempo (ABC) aumentan de forma aproximadamente proporcional a la dosis en el intervalo de dosis terapéuticas. Poblacionesespeciales: Pacientes geriátricos (mayores de 65 años): En individuos sanos de edad avanzada (≥ 70 años) la exposición general a GALVUS® (100 mg/día) fue 32% más elevada (con un aumento del 18% en la concentración plasmática máxima) que la de los individuos sanos más jóvenes (18-40 años). Estas diferencias carecen de interés clínico. En los grupos etarios estudiados, la edad no afectaba la inhibición de DDP-4 con GALVUS®. Pacientes pediátricos (menores de 18 años): No se dispone de datos farmacocinéticos para esta población de pacientes. Grupo étnico: No se tienen pruebas de que el grupo étnico afecte la farmacocinética de GALVUS®. Género: No se observaron diferencias en la farmacocinética de GALVUS® entre individuos de sexo masculino y femenino de edades diferentes y con índices de masa corporal (IMC) diversos. El género no afecta la inhibición de DPP4 con GALVUS®. Obesidad: El IMC no mostró impacto en los parámetros farmacocinéticos de GALVUS®. La inhibición de GALVUS® en la DPP4 no fue afectada por el IMC. Insuficiencia hepática: Se estudió el efecto de la insuficiencia hepática en la farmacocinética de GALVUS® en individuos con insuficiencia hepática leve, moderada y severa tomando como base la escala Child-Pugh (puntuación de 6 para los casos leves hasta 12 para los casos graves), en comparación con individuos con función hepática normal. Tras la administración de una sola dosis, la exposición a GALVUS® (100 mg) disminuyó 20% y 8% en individuos que padecían insuficiencia hepática leve o moderada, respectivamente, pero aumentaba un 22% en los que padecían insuficiencia grave. El máximo cambio (aumento o disminución) de exposición a GALVUS® fue ~30%, el cual no se considera clínicamente relevante. No hubo correlación alguna entre la gravedad de la insuficiencia hepática y los cambios en la exposición a GALVUS®. El uso de GALVUS® no está recomendado en pacientes con insuficiencia hepática, incluyendo pacientes que presenten enzimas hepáticas (ALT o AST) > 2.5 el límite superior normal antes de iniciar tratamiento. Insuficiencia renal: En comparación con sujetos sanos normales, el ABC de la vildagliptina es en promedio 1.4, 1.7 y 2 veces mayor en pacientes con disfunción renal leve, moderada o grave, respectivamente. En comparación con voluntarios sanos, el ABC de los metabolitos LAY151 y BQS867 es en promedio, respectivamente unas 1.6, 3.2 y 7.3 y unas 1.4, 2.7 y 7.3 veces mayor en pacientes con disfunción renal leve, moderada o grave. Los escasos datos obtenidos en pacientes con nefropatía terminal indican que, en esos pacientes, la exposición a la vildagliptina es similar a la de los pacientes con insuficiencia renal grave. La concentración de LAY151 en pacientes con nefropatía terminal era aproximadamente 2-3 veces mayor que la observada en pacientes con disfunción renal grave. Puede que sea necesario ajustar la dosis en los pacientes con disfunción renal (véase el apartado Dosis y vía de administración). La vildagliptina se elimina por hemodiálisis en grado limitado (3% durante una sesión de hemodiálisis de 3-4 horas iniciada 4 horas después de la administración de la dosis). Farmacodinamia: La administración de vildagliptina inhibe rápida y completamente la actividad de la DPP4. En los pacientes con diabetes de tipo 2, la vildagliptina inhibió la actividad de la DPP4 durante 24 horas. La vildagliptina, al incrementar las concentraciones endógenas de dichas hormonas incretinas, potencia la sensibilidad de las células beta a la glucosa resultando en mejor secreción de insulina dependiente de glucosa. El tratamiento con 500 a 100 mg al día en pacientes con Diabetes Mellitus tipo 2 significativamente mejora los marcadores de función de las células beta. El grado de mejora en la función de las células beta es dependiente en la etapa inicial de mejora; en individuos no diabéticos (glucemia normal), la vildagliptina no estimula la secreción de insulina o reduce los niveles de glucosa. Al incrementar los niveles endógenos de GLP-1, la vildagliptina mejora la sensibilidad de las células alfa a la glucosa, resultando en mayor secreción de glucagón apropiado de Glucosa. La reducción en glucagón inapropiado durante las comidas, por el contrario, atenúa la resistencia a insulina. El incremento mejorado en la proporción de insulina/glucagón durante la hiperglucemia debido al incremento en los niveles de la hormona incretina resulta en disminución en la producción hepática de glucosa en ayuno y post prandial, llevando a glucemia reducida. No se observa con el tratamiento de vildagliptina el conocido efecto de los niveles incrementados de GLP-1 para reducir el tiempo de vaciado gástrico no es observado con el tratamiento de vildagliptina. Adicionalmente, se ha observado reducción en la lipemia postprandial que no está asociada con el efecto mediado por vildagliptina en las incretinas para mejorar la función del islote.
Contraindicaciones: GALVUS® está contraindicado en pacientes con hipersensibilidad conocida a la vildagliptina o a cualquiera de los excipientes.
Precauciones generales: General: GALVUS® no es un sustituto de la insulina en pacientes que necesitan insulina. No se debe utilizar GALVUS® en pacientes con diabetes tipo 1 o para el tratamiento de la cetoacidosis diabética. Alteración de la función hepática: No se recomienda el uso de GALVUS® en pacientes con disfunción hepática, incluyendo pacientes con enzimas hepáticas (ALT o AST) > 2.5 el límite superior normal previo al inicio de tratamiento. Monitoreo de enzimas hepáticas: Se han reportado casos raros de disfunción hepática (incluyendo hepatitis). En estos casos los pacientes se presentaron generalmente asintomáticos, sin secuelas clínicas y en las pruebas de función hepática (PFH"s) regresaron a la normalidad después de descontinuar el tratamiento. Se deben realizar PFH"s antes de iniciar tratamiento con GALVUS®. Se deben monitorear las PFH"s durante el tratamiento con GALVUS® cada 3 meses durante el primer año y después de manera periódica. Los pacientes que desarrollen aumento de los niveles de transaminasas deben ser monitoreados con una segunda evaluación de las PFH"s para confirmar el hallazgo, y se debe dar seguimiento regular con evaluación de las PFH"s hasta que la anormalidad(es) se normalicen. Si persiste el incremento de AST o ALT 3 veces del límite superior normal se recomienda suspender el tratamiento con GALVUS®. Los pacientes que desarrollen ictericia u otros signos sugestivos de disfunción hepática deben descontinuar el tratamiento con GALVUS® y contactar a su médico de forma inmediata. Una vez que las PFH"s se normalicen posterior a la suspensión del tratamiento con GALVUS®, no se debe reiniciar el tratamiento con GALVUS®. Insuficiencia cardiaca: Un estudio clínico de la vildagliptina efectuado en pacientes con insuficiencia cardiaca de las clases funcionales I-III de la NYHA (Asociación del Corazón de Nueva York, New York Heart Association por sus siglas en inglés) reveló que el tratamiento con vildagliptina no se asociaba a una alteración del funcionamiento del ventrículo izquierdo ni a un agravamiento de la insuficiencia cardiaca congestiva (ICC) preexistente en comparación con el placebo. La experiencia clínica en pacientes de la clase funcional III de la NYHA tratados con vildagliptina sigue siendo escasa y los resultados no son concluyentes. No hay experiencia del uso de vildagliptina en estudios clínicos en pacientes clasificados como NYHA grado IV y por lo tanto no se recomienda el uso de GALVUS® en estos pacientes.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: La vildagliptina no fue teratógena en la rata ni en el conejo. No obstante, como no se tiene suficiente experiencia de uso de GALVUS® en mujeres embarazadas, GALVUS® no debe usarse durante el embarazo, a menos que los beneficios para la madre justifiquen el riesgo para el feto. Lactancia: GALVUS® no debe administrarse a madres lactantes, pues no se sabe si la vildagliptina pasa a la leche humana. Fertilidad: No se han realizado estudios sobre la fertilidad humana con GALVUS®. Los estudios de fertilidad realizados en ratas con dosis de vildagliptina hasta 200 veces mayores que la dosis humana recomendada no han revelado una degradación de la fertilidad o del desarrollo embrionario inicial.
Reacciones secundarias y adversas: Resumen del perfil de seguridad: La seguridad y la tolerabilidad de la vildagliptina (50 mg al día y 50 mg dos veces al día y 100 mg una vez al día) se han evaluado agrupando los datos de más de 11,000 pacientes que participaron en 36 estudios de fase II y III (incluidos 3 sin enmascaramiento) de 12 a más de 104 semanas de duración. En tales estudios, la vildagliptina se administró en monoterapia, como tratamiento aditivo a otros antidiabéticos orales (metformina, una TZD, una SU e insulina) o en terapia doble inicial con metformina o pioglitazona. Los pacientes de los grupos de comparación recibieron ya sea el placebo solo o bien metformina, TZD, SU, acarbosa o insulina. Para calcular la frecuencia de las reacciones adversas observadas en cada indicación se tuvieron en cuenta los datos de seguridad de un subgrupo de estudios comparativos fundamentales que duraron por lo menos 12 semanas. Los datos de seguridad procedían de pacientes expuestos a una dosis diaria de vildagliptina de 50 mg (una vez al día) o 100 mg (50 mg dos veces al día o 100 mg una vez al día), que se administró sola o asociada a otro fármaco. En dichos estudios, la mayoría de las reacciones adversas fueron leves y transitorias, sin requerir descontinuación del tratamiento. No se encontró correlación entre los eventos adversos y la edad, sexo, grupo étnico, duración a la exposición o la dosis diaria. Se han reportado casos raros de angioedema en los grupos de vildagliptina en incidencia similar a los grupos control, una mayor proporción de casos fue reportada con vildagliptina cuando se administró en combinación con inhibidores de la enzima convertidora de angiotensina (IECA). La mayoría de estos eventos fueron leves y se resolvieron sin interrumpir el tratamiento con vildagliptina. Se han reportado raramente casos de disfunción hepática (incluyendo hepatitis). En estos casos los pacientes cursaron asintomáticos sin secuelas clínicas y las PFH"s regresaron a lo normal después de descontinuar el tratamiento. En los estudios controlados de monoterapia y de adición hasta 24 semanas de duración la incidencia de elevación de ALT o AST mayor o igual a 3 x el límite superior normal (presente en por lo menos 2 mediciones consecutivas o en la visita final de tratamiento) fue de 0.2%, 0.3% y 0.2% para vildagliptina 50 mg al día, vildagliptina 50 mg dos veces al día y todos los comparadores respectivamente. Estas elevaciones en transaminasas fueron generalmente asintomáticas, de naturaleza no progresiva y no se asociaron con colestasis o ictericia. Resumen tabulado de reacciones adversas descritas en estudios clínicos: A continuación se indican las reacciones adversas registradas en pacientes que recibieron GALVUS® en monoterapia o como tratamiento aditivo en estudios con doble enmascaramiento, ordenadas por clase de órgano, aparato o sistema (MedDRA), y por frecuencia absoluta. Dentro de cada clase de órgano, aparato o sistema, las reacciones se clasifican por orden decreciente de frecuencia. En cada grupo de frecuencia, las reacciones se especifican por orden decreciente de gravedad. También se indica la categoría de frecuencia de cada reacción adversa aplicando la siguiente convención (CIOMS III): muy frecuentes (≥ 1/10); frecuentes (≥ 1/100 a < 1/10); infrecuentes (≥ 1/1000 a < 1/100); raras (≥ 1/10 000 a < 1/1000); muy raras ( < 1/10 000). Monoterapia: La incidencia total de descontinuaciones de los estudios de monoterapia debido a eventos adversos no fue mayor en pacientes tratados con vildagliptina a dosis de 50 mg una vez al día (0.2%), o vildagliptina a dosis de 50 mg dos veces al día (0.1%) que en el grupo placebo (0.6%) o los comparadores (0.5%). En los estudios de monoterapia con controles de referencia, la hipoglucemia fue una reacción adversa ocasional en los pacientes que recibieron GALVUS® en monoterapia a dosis de 50 mg una vez al día (0.5%, 2 de 409), y en dosis de 50 mg dos veces al día (0.3%, 4 de 1,373) en comparación con los pacientes en los grupos placebo y con comparador activo (0.2%, 2 de 1,082). Sin reporte de eventos adversos serios o severos. GALVUS® no demostró aumento de peso cuando se administra como monoterapia. Tabla 1. Reacciones adversas notificadas en pacientes que recibieron GALVUS® a dosis de 50 mg una vez al día (n = 409 o 50 mg dos veces al día n = 1,373) en monoterapia en los estudios con doble ciego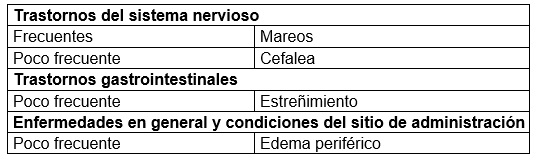
Los estudios clínicos a largo plazo hasta 2 años no muestran señales adicionales de riesgos no esperados con vildagliptina en monoterapia. Combinación con metformina: En los estudios clínicos de combinación de vildagliptina + metformina, 0.4% de los pacientes fueron descontinuados debido a eventos adversos en el grupo de vildagliptina 50 mg una vez al día + metformina y no se reportaron descontinuaciones por eventos adversos en los grupos de vildagliptina 50 mg dos veces al día + metformina o placebo más metformina. En los estudios de tratamiento complementario con controles de referencia, la incidencia de hipoglucemia en los pacientes que recibieron vildagliptina asociada con metformina fue ocasional en el grupo de vildagliptina 50 mg una vez al día en combinación con metformina (0.9%), vildagliptina 50 mg dos veces al día en combinación con metformina (0.5%) y en el grupo placebo + metformina (0.4%). No se reportaron eventos de hipoglucemia severa en los grupos de vildagliptina. GALVUS® no demostró aumento de peso cuando fue administrada en combinación con metformina. Tabla 2. Reacciones adversas notificadas en pacientes que recibieron GALVUS® 50 mg una vez al día (n = 233) o 50 mg dos veces al día (n = 183) en combinación con metformina en los estudios con doble ciego
Los estudios clínicos a largo plazo de hasta 2 años de duración no revelaron nuevas cuestiones de seguridad ni riesgos imprevistos al añadir vildagliptina al tratamiento con metformina. Al investigar vildagliptina en asociación con metformina para el tratamiento inicial, no se observaron nuevas cuestiones de seguridad ni riesgos imprevistos. Combinación con sulfonilureas: En los estudios clínicos de combinación de vildagliptina 50 mg + Glimepirida, la incidencia total de descontinuaciones debido a eventos adversos fue de 0.6% en el grupo de vildagliptina 50 mg + Glimepirida en comparación con 0% en grupo placebo + Glimepirida. En los estudios clínicos la incidencia de hipoglicemia cuando se adicionó vildagliptina 50 mg una vez al día fue de 1.2% en comparación con 0.6% para el grupo de placebo + Glimepirida. No se reportaron eventos severos de hipoglucemia en los grupos de vildagliptina. A la dosis recomendada de 50 mg, GALVUS® no demostró aumento de peso al ser administrado en combinación con Glimepirida. Tabla 3. Reacciones adversas notificadas en pacientes que recibieron GALVUS® 50 mg una vez al día en combinación con una sulfonilurea en los estudios con doble ciego (n = 170)
Combinación con tiazolidinedionas (TZD): En estudios clínicos con la combinación de vildagliptina y una TZD, 0.7% de los pacientes fueron descontinuados por eventos adversos en el grupo de vildagliptina 50 mg una vez al día + Pioglitazona, y no se reportaron descontinuaciones debido a eventos adversos en los grupos de vildagliptina 50 mg dos veces al día + Pioglitazona o placebo + Pioglitazona. En los estudios clínicos no se reportaron eventos de hipoglucemia en los pacientes que recibieron vildagliptina 50 mg una vez al día más Pioglitazona 45 mg; la hipoglucemia fue ocasional en los pacientes que recibieron vildagliptina 50 mg dos veces al día más Pioglitazona 45 mg (0.6%), y frecuente en los pacientes que recibieron placebo + Pioglitazona 45 mg (1.9%). No se reportaron eventos hipoglucémicos severos en los grupos con vildagliptina. En el estudio de adición a Pioglitazona el cambio de peso corporal en comparación con placebo fue de +0.1 Kg y +1.3 Kg para 50 mg una vez al día y 50 mg dos veces al día respectivamente. La incidencia de edema periférico cuando vildagliptina se adicionó a la dosis máxima de Pioglitazona (45 mg una vez al día) fue de 8.2% para el grupo de 50 mg una vez al día, y de 7.0% para el grupo de 50 mg dos veces al día en comparación con 2.5% para el grupo Pioglitazona. La incidencia de edema cuando vildagliptina adicionada a Pioglitazona como terapia combinada inicial en pacientes sin tratamiento farmacológico previo fue menor que para monoterapia con Pioglitazona (50 mg una vez al día 3.5%, 50 mg dos veces al día 6.1%, Pioglitazona 30 mg 9.3%). Tabla 4. Reacciones adversas notificadas en pacientes que recibieron GALVUS® a dosis de 50 mg una vez al día (n = 146 o 50 mg dos veces al día n = 158) en combinación con una TZD en los estudios con doble-ciego
Combinación con insulina: En los estudios clínicos comparativos en los que se administró 50 mg de vildagliptina dos veces al día asociada a la insulina (con o sin metformina), la incidencia total de abandonos debido a reacciones adversas fue del 0.3% en el grupo de la vildagliptina y no se registraron retiradas en el grupo del placebo. En ambos grupos terapéuticos se registró una incidencia similar de hipoglucemia (14.0% en el grupo de la vildagliptina y 16.4% en el del placebo). Dos pacientes del grupo de la vildagliptina y 6 del grupo del placebo refirieron episodios de hipoglucemia grave. Al final del estudio, el efecto sobre el peso corporal medio fue neutro (hubo una diferencia de peso de + 0.6 kg en el grupo de la vildagliptina y ninguna variación de peso en el grupo del placebo). Tabla 5. Reacciones adversas notificadas en pacientes que recibieron GALVUS® a dosis de 50 mg dos veces al día (con o sin metformina [n = 371])
Triterapia con metformina y una SU: No se notificaron abandonos por reacciones adversas en el grupo de la vildagliptina + metformina + glimepirida; en cambio, se notificó un 0.6% de abandonos en el grupo del placebo + metformina + glimepirida. En ambos grupos terapéuticos la hipoglucemia fue frecuente (se registró un 5.1% de casos en el grupo de la vildagliptina + metformina + glimepirida y un 1.9% de casos en el grupo del placebo + metformina + glimepirida). En el grupo de la vildagliptina se registró un episodio grave de hipoglucemia. Al final del estudio, el efecto sobre el peso corporal medio fue neutro (hubo una diferencia de peso de +0.6 kg en el grupo de la vildagliptina y de -0.1 kg en el grupo del placebo). Tabla 6. Reacciones adversas descritas en pacientes que recibieron 50 mg de GALVUS® dos veces al día asociado a la metformina y una SU (n = 157)
Reacciones adversas en notificaciones espontáneas y casos publicados (de frecuencia desconocida): Desde la comercialización de GALVUS®, se han descrito las reacciones adversas siguientes a través de comunicaciones espontáneas de casos y de casos publicados en la literatura específica. Como dichas reacciones las comunica de forma voluntaria una población de tamaño incierto no siempre es posible estimar de forma fiable su frecuencia, de modo que esta última se considera de frecuencia desconocida. Hepatitis reversible tras el retiro del medicamento (ver Precauciones generales). Urticaria, lesiones cutáneas ampollosas y exfoliativas, incluyendo Pénfigo bulloso. [75] Artralgia, en ocasiones de carácter severo.
Interacciones medicamentosas y de otro género: El potencial de interacción farmacológica de vildagliptina es reducido. La vildagliptina no es un sustrato del citocromo P450 (CYP), ni tampoco inhibe ni induce las enzimas del CYP450, de modo que es improbable que interaccione con fármacos que son sustratos, inhibidores o inductores de dichas enzimas. Además, la vildagliptina no afecta la depuración metabólica de fármacos metabolizados por las enzimas CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 y CYP3A4/5. Se han realizado estudios de interacción farmacológica con los medicamentos comúnmente prescritos a pacientes con diabetes tipo 2 y con medicamentos cuyo margen de seguridad es estrecho. Los resultados de dichos estudios no han revelado interacciones clínicamente significativas con otros antidiabéticos orales (glibenclamida, pioglitazona, metformina), amlodipino, digoxina, ramipril, simvastatina, valsartán o warfarina, cuando estos fármacos se coadministran con vildagliptina.
Alteraciones en los resultados de pruebas de laboratorio: Monitoreo de enzimas hepáticas: Se han reportado casos raros de disfunción hepática (incluyendo hepatitis). En estos casos los pacientes se presentaron generalmente asintomáticos, sin secuelas clínicas y en las pruebas de función hepática (PFH"s) regresaron a la normalidad después de descontinuar el tratamiento. Se deben realizar PFH"s antes de iniciar tratamiento con GALVUS®. Se deben monitorear las PFH"s durante el tratamiento con GALVUS® cada 3 meses durante el primer año y después de manera periódica. Los pacientes que desarrollen aumento de los niveles de transaminasas deben ser monitoreados con una segunda evaluación de las PFH"s para confirmar el hallazgo, y se debe dar seguimiento regular con evaluación de las PFH"s hasta que la anormalidad(es) se normalicen. Si persiste el incremento de AST o ALT 3 veces más del límite superior normal se recomienda suspender el tratamiento con GALVUS®. Los pacientes que desarrollen ictericia u otros signos sugestivos de disfunción hepática deben descontinuar el tratamiento con GALVUS® y contactar a su médico de forma inmediata. Una vez que las PFH"s se normalicen posterior a la suspensión del tratamiento con GALVUS®, no se debe reiniciar el tratamiento con GALVUS®.
Precauciones y relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Un estudio de carcinogenicidad fue realizado en ratas a dosis orales hasta 900 mg/kg (aproximadamente 200 veces la exposición en humanos a la dosis máxima recomendada). No se observó incremento en la incidencia de tumores atribuible a la vildagliptina. Un estudio de carcinogenicidad a dos años fue realizado en ratones a dosis orales hasta 1,000 mg/kg (hasta 240 veces la exposición en humanos a la dosis máxima recomendada). La incidencia de tumores mamarios fue incrementada en ratones hembra a aproximadamente 150 veces la exposición máxima a la vildagliptina anticipada en humanos; no hubo incremento a 60 veces la exposición máxima en humanos. La incidencia de hemoangiosarcoma fue incrementada en ratones machos tratados de 42 a 240 veces la exposición máxima en humanos a la vildagliptina y en ratones hembra a 150 veces la exposición máxima en humanos. No se encontró incremento significativo en la incidencia de hemoangiosarcoma a aproximadamente 16 veces la exposición máxima en humanos a la vildagliptina en machos y aproximadamente 60 veces la exposición máxima en hembras. La vildagliptina no fue mutagénica en diversas pruebas mutagénicas incluyendo la prueba de mutación bacteriana inversa de Ames y el estudio de aberración cromosómica de linfocitos humanos. La prueba de micronúcleos de células de médula en ratas y ratones no reveló potencial clastogénico y aneugénico hasta 2,000 mg/Kg o aproximadamente 400 veces la exposición máxima en humanos. Utilizando esta misma dosis, también fue negativa la microelectroforesis de células individuales de hígado de ratón in vivo (ensayo "cometa"). En un estudio de toxicología de 13 semanas en monos se observaron lesiones en la piel a dosis mayores a 5 mg/Kg/día. Estas lesiones se localizaban en las extremidades (manos, pies, orejas y cola). A dosis de 5 mg/kg/día (el equivalente aproximado a la dosis de 100 mg en humanos), sólo se observaron ampollas. Éstas residieron a pesar de que se continuó el tratamiento y no se asociaron anormalidades histopatológicas. Descamación de la piel, costras y llagas en la cola con cambios histopatológicos asociados fueron observados a dosis mayores a 20 mg/kg/día (aproximadamente 3 veces mayor a la dosis de 100 mg en humanos). Lesiones necróticas de la cola se observaron a dosis mayores a 80 mg/kg/día. Es de notar que vildagliptina manifiesta una potencia farmacológica mayor en monos en comparación con humanos. Las lesiones de la piel no fueron reversibles en los monos tratados a dosis de 160 mg/kg/día durante un periodo de recuperación de 4 semanas. No se han observado lesiones en la piel en otras especies animales ni en humanos tratados con vildagliptina.
Dosis y vía de administración: Dosis: La administración del tratamiento contra la diabetes debe adaptarse a las necesidades del individuo. La dosis recomendada de GALVUS® es de 50 mg una o dos veces al día. La dosis máxima de GALVUS® es de 100 mg al día. En monoterapia, en combinación con metformina, con TZD o con insulina (con o sin metformina), la dosis recomendada de GALVUS® es de 50 mg o 100 mg al día. En combinación con una SU la dosis recomendada es de 50 mg al día; en esta población de pacientes la administración de vildagliptina 100 mg al día no demostró mayor efectividad que vildagliptina 50 mg al día. En la terapia triple con metformina y una SU, la dosis recomendada de GALVUS® es de 100 mg al día. Si fuera necesario un control glucémico reforzado, además de la dosis diaria recomendada máxima de 100 mg se pueden utilizar adicionalmente otros antidiabéticos, como la metformina, una SU, una TZD o de insulina. Población general destinataria: Pacientes mayores de 18 años de edad. Poblaciones especiales: Pacientes con insuficiencia renal: No es preciso ajustar la dosis de GALVUS® en pacientes con insuficiencia renal leve. En los pacientes con insuficiencia renal moderada o grave o con nefropatía terminal sometidos a hemodiálisis la dosis recomendada de GALVUS® es de 50 mg una vez al día (ver Farmacocinética). Pacientes con insuficiencia hepática: GALVUS® no se recomienda en pacientes con disfunción hepática incluyendo pacientes con una ALT o AST > 2.5% x el límite superior normal previo a iniciar tratamiento (ver Farmacocinética). Pacientes pediátricos (menores de 18 años): No se han estudiado los efectos de GALVUS® en pacientes menores de 18 años; por consiguiente, no se recomienda la utilización de este medicamento en pacientes pediátricos (ver Farmacocinética). Pacientes geriátricos (65 años o más): En pacientes ≥ 65 años y ≥ 75 años tratados con GALVUS®, no se observaron diferencias de eficacia, tolerabilidad o seguridad general con respecto a pacientes más jóvenes. Por lo tanto, no es necesario ajustar la dosis en pacientes de edad avanzada (ver Farmacocinética). Vía de administración: Oral. Método de administración: GALVUS® puede administrarse con o sin alimentos. La dosis de 50 mg una vez al día se debe administrar por la mañana. La dosis de 100 mg se debe administrar en dos tomas separadas de 50 mg cada una, por la mañana y por la noche. Si se olvida una dosis de GALVUS®, debe de administrarse tan pronto el paciente se acuerde. No debe de usarse una dosis doble en el mismo día.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: Signos y síntomas: GALVUS® se administró en dosis de 25, 50, 100, 200, 400 y 600 mg una vez al día a individuos sanos (siete a catorce individuos por grupo de tratamiento) durante 10 días consecutivos. Se toleraron bien dosis de hasta 200 mg. A la dosis de 400 mg hubo tres casos de dolor muscular y casos aislados de parestesia leve y transitoria, fiebre, edema y alzas transitorias de la concentración de lipasa (el doble del máximo normal). A la dosis de 600 mg un individuo experimentó edema en pies y manos, un aumento exagerado de la concentración de creatina-fosfocinasa y aumentos concomitantes de la concentración de alanina-aminotransferasa (AST), proteína C-reactiva y mioglobina. Otras tres personas del mismo grupo posológico presentaron edemas en ambos pies, acompañados de parestesia en dos de ellas. Todos los síntomas y anomalías de laboratorio se resolvieron al suspender la administración del fármaco de estudio. Tratamiento: GALVUS® no es dializable, pero el principal metabolito de la hidrólisis (LAY151) se puede eliminar por hemodiálisis.
Presentaciones: Caja con 7, 14, 28, 30 o 56 comprimidos de 50 mg.
Recomendaciones sobre almacenamiento: Consérvese a no más de 30°C. Consérvese la caja bien cerrada.
Leyendas de protección: Su venta requiere receta médica. No se deje al alcance de los niños. No se use en el embarazo, ni lactancia. Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx
Nombre y domicilio del laboratorio: Propiedad de: Novartis Pharma AG. Lichtstrasse 35, 4056 Basel, Suiza Representante legal e importador: NOVARTIS FARMACÉUTICA, S.A. de C.V. Calz. de Tlalpan No. 1779 Col. San Diego Churubusco, C.P. 04120 Deleg. Coyoacán, Ciudad de México, México
Número de registro del medicamento: 045M2007, SSA IV