HERCEPTIN®
ROCHE
Denominación genérica: Trastuzumab.
Forma farmacéutica y formulación: HERCEPTIN® Vial multidosis de 440 mg. Solución. El frasco ámpula con polvo contiene: trastuzumab 440mg. Excipiente cbp. El frasco ámpula con diluyente contiene: agua inyectable 20ml.
Indicaciones terapéuticas: Cáncer de mama metastásico (CMM): HERCEPTIN® está indicado para el tratamiento del cáncer de mama metastásico con sobreexpresión de la proteína HER2: como monoterapia para el tratamiento de las pacientes que han recibido uno o más regímenes quimioterapéuticos para la enfermedad metastásica. En combinación con paclitaxel o docetaxel para el tratamiento de las pacientes que no han recibido quimioterapia previa para la enfermedad metastásica. En combinación con un inhibidor de aromatasa para el tratamiento de pacientes con cáncer de mama metastásico positivo a receptores hormonales. Cáncer de mama temprano (CMT): HERCEPTIN® está indicado para el tratamiento del cáncer de mama temprano HER2 positivo tras la cirugía, la quimioterapia (neoadyuvante o adyuvante) y/o la radioterapia (si procede).
Farmacocinética y farmacodinamia: Farmacocinética: se ha estudiado la farmacocinética de trastuzumab en pacientes con cáncer de mama metastásico y cáncer de mama temprano. En los estudios de fase I, la farmacocinética dependiente de la dosis quedó demostrada por infusiones intravenosas de corta duración de 10, 50, 100, 250 y 500 mg de trastuzumab una vez a la semana, observándose que al incrementar la dosis, aumenta también la vida media promedio y disminuye la depuración renal. Farmacocinética en estado de equilibrio: para estimar la farmacocinética en estado de equilibrio en pacientes tratados con trastuzumab en una dosis de carga de 4 mg/kg seguida de una dosis de mantenimiento de 2 mg/kg/semana, se aplicó un método de farmacocinética poblacional, utilizando datos obtenidos en estudios de fase I, fase II y estudios principales de fase III. En esta evaluación, la depuración típica de trastuzumab fue de 0,225 l/día; el volumen de distribución típico, de 2,95 l, y la vida media terminal, de 28,5 días (intervalo de confianza del 95%, 25,5-32,8 días). Al cabo de 143 días, o aproximadamente 20 semanas, el ABC en estado de equilibrio de una semana sería de 578 mg/día/l, con una concentración máxima y mínima de 110 mg/l y 66 mg/l, respectivamente. El mismo intervalo de tiempo correspondería a la eliminación del trastuzumab tras la suspensión del tratamiento con HERCEPTIN®. Se utilizó un método farmacocinético para la población, utilizando los datos de los estudios de fase I, de fase II y los estudios pivote de fase III, para estimar la farmacocinética en estado estable en las pacientes a quienes se les administró trastuzumab a una dosis de carga de 4 mg/kg, seguida por una dosis de mantenimiento semanal de 2 mg/kg. En esta evaluación, la depuración típica de trastuzumab fue de 0,225 l/día y el volumen de distribución típico fue de 2,95 l, con una correspondiente vida media terminal de 28,5 días (intervalo de confianza del 95%, 25,5-32,8 días). El ABC semanal en estado estable de 578 mg/día/l, las concentraciones máximas de 110 mg/l y las concentraciones estables de 66 mg/l, deben alcanzarse en 143 días o aproximadamente 20 semanas. El mismo intervalo de tiempo podría predecirse para la eliminación de trastuzumab después de suspender la terapia con HERCEPTIN®. En las pacientes con cáncer de mama temprano tratadas con HERCEPTIN® en una dosis de carga de 8 mg/kg seguida de una dosis de mantenimiento de 6 mg/kg cada tres semanas se alcanzaron concentraciones mínimas en estado de equilibrio de 63 mg/l, en el ciclo 13. Estas concentraciones eran comparables con las notificadas anteriormente en pacientes con cáncer de mama metastásico. La administración de la quimioterapia concomitante (con antraciclinas/ciclofosfamida, paclitaxel o docetaxel) no pareció influir en la farmacocinética de trastuzumab. La administración de anastrozole concomitante no tuvo influencia en la farmacocinética de trastuzumab. Farmacocinética en poblaciones especiales: no se han realizado estudios de farmacocinética detallados en personas ancianas y en pacientes con insuficiencia renal o hepática. Ancianos: se ha demostrado que la edad no influye en la distribución y la eliminación de trastuzumab (ver Dosis y vía de administración). Farmacodinamia: mecanismo de acción: trastuzumab es un anticuerpo monoclonal derivado de ADN recombinante humano, dirigido de forma selectiva al sitio de acción extracelular de la proteína del receptor 2 del factor de crecimiento epidérmico humano (HER2). Este anticuerpo es una IgG1 que contiene regiones estructurales humanas que se complementan con las regiones determinantes de un anticuerpo anti-p185-HER2 murino que se enlaza al receptor HER2. El proto-oncogen HER2 o cerbB2 codifica para una única proteína transmembranal receptora de 185 kDa, que se encuentra estructuralmente relacionada con el receptor del factor de crecimiento epidérmico. En un 25-30% de los cánceres de mama primarios, se ha encontrado sobreexpresión del gen HER2. Una consecuencia de la amplificación del gen HER2 es un incremento en la expresión de la proteína HER2 en la superficie de las células tumorales, lo cual da como resultado un receptor HER2 constitutivamente activo. Los estudios indican que las pacientes cuyos tumores presentan una amplificación o sobreexpresión de HER2 tienen una supervivencia, libre de enfermedad, más corta que la de aquellos que presentan tumores sin amplificación o sobreexpresión de HER2. Tanto en los estudios in vitro como en la experimentación animal, se ha comprobado que el trastuzumab inhibe la proliferación de las células tumorales humanas con sobreexpresión de HER2. Se ha demostrado in vitro que la citotoxicidad celular dependiente de anticuerpos (ADCC) mediada por el trastuzumab afecta más a las células cancerosas con sobreexpresión de HER2 que a las células cancerosas sin sobreexpresión de HER2. Eficacia: cáncer de mama metastásico (CMM): en los ensayos clínicos, HERCEPTIN® se ha utilizado en régimen monoterapéutico en pacientes con cáncer de mama metastásico y sobreexpresión de HER2, y que habían recaído tras una o más pautas quimioterapéuticas contra la enfermedad metastásica. También se ha utilizado HERCEPTIN® en los ensayos clínicos asociado a paclitaxel o una antraciclina (doxorrubicina o epirrubicina) más ciclofosfamida (A y C) como tratamiento de primera línea en pacientes con cáncer de mama metastásico con sobreexpresión de HER2. Las pacientes que habían recibido previamente tratamiento adyuvante con antraciclinas, recibieron paclitaxel (175 mg/m2 en infusión de 3 horas) con o sin HERCEPTIN®. Las pacientes pudieron ser tratadas con HERCEPTIN® hasta la progresión de su enfermedad. Se ha obtenido una tasa de respuesta tumoral global del 15% y una supervivencia media de 13 meses, al utilizar HERCEPTIN® en régimen monoterapéutico como tratamiento de segunda o tercera línea en mujeres con cáncer de mama metastásico y sobreexpresión de HER2. El uso de HERCEPTIN® en combinación con paclitaxel como tratamiento de primera línea en mujeres con cáncer de mama metastásico con sobreexpresión de HER2 prolonga significativamente el tiempo promedio de la progresión de la enfermedad, en comparación con las pacientes que reciben el tratamiento con paclitaxel solamente. El incremento del tiempo promedio en la progresión de la enfermedad de las pacientes tratadas con paclitaxel es de 3,9 meses (6,9 meses versus 3,0 meses). La respuesta tumoral y la tasa de supervivencia al cabo de un año también se incrementaron para la combinación de HERCEPTIN® y paclitaxel respecto de paclitaxel solo. En un ensayo clínico controlado y aleatorizado, también se ha estudiado HERCEPTIN® en asociación con docetaxel como tratamiento de primera línea de mujeres con cáncer de mama metastásico. La asociación de HERCEPTIN® y docetaxel elevó significativamente la tasa de respuesta (61% versus 34%) y prolongó el tiempo promedio de la progresión de la enfermedad (en 5,6 meses) en comparación con las pacientes tratadas con docetaxel solo. En comparación con docetaxel en monoterapia, el tiempo promedio de la supervivencia también aumentó significativamente con esta asociación (31,2 versus 22,7 meses). Tratamiento combinado con HERCEPTIN® y anastrozole: se ha estudiado HERCEPTIN® en combinación con anastrozole para tratamiento de primera línea de cáncer de mama metastásico en pacientes con sobreexpresión de HER2, positivos a receptores hormonales (p. ej., receptores a estrógenos (ER) y/o receptores a progesterona (PR)). El tiempo de supervivencia libre de progresión de enfermedad se duplicó en el brazo del estudio de HERCEPTIN® más anastrozole (4,8 meses contra 2,4 meses) en comparación con anastrozole solo. Para los otros parámetros, la mejora observada para la combinación fue: respuesta global (16,5% contra 6,7%), tasa de beneficio clínico (42,7% contra 27,9%); tiempo hasta la progresión (4,8 meses contra 2,4 meses). Para el tiempo de respuesta y duración de respuesta, no se pudo registrar una diferencia entre ambos brazos. La supervivencia media global se extendió por 4,6 meses para pacientes en el brazo con tratamiento combinado. La diferencia no fue estadísticamente significativa; sin embargo, más de la mitad de los pacientes en el brazo de anastrozole solo cruzaron al régimen con HERCEPTIN® después de la progresión de la enfermedad. Cincuenta y dos por ciento de los pacientes en tratamiento con HERCEPTIN® más anastrozole sobrevivieron por al menos 2 años, en comparación con 45% en tratamiento de anastrozole solo. Cáncer de mama temprano (CMT): como tratamiento adyuvante, HERCEPTIN® se ha estudiado en un ensayo clínico (HERA) multicéntrico y aleatorizado para comparar un año de administración de HERCEPTIN® cada tres semanas con sólo observación en pacientes que sufrían cáncer de mama temprano HER2 positivo tras cirugía, quimioterapia convencional y radioterapia (si procedía). A las pacientes asignadas al grupo de HERCEPTIN® se les administró una dosis de carga de 8 mg/kg, seguida de 6 mg/kg cada tres semanas durante un año.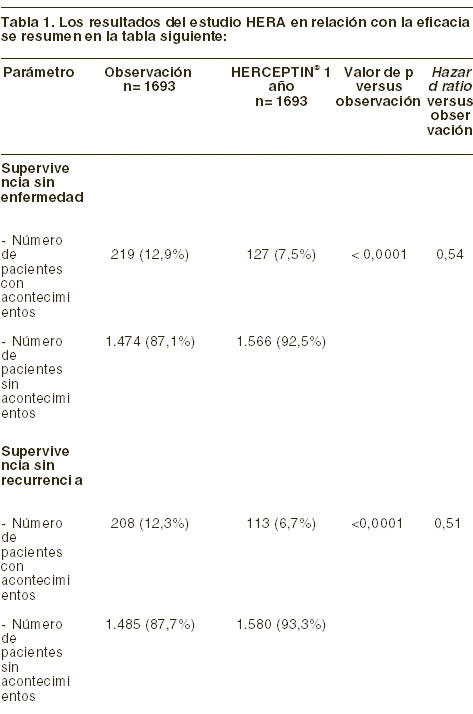
En la variable de evaluación principal -la supervivencia sin enfermedad-, el cociente de riesgo instantáneo (hazard ratio) muestra un beneficio absoluto, en términos de supervivencia sin enfermedad a los 2 años, de 7,6 puntos porcentuales (85,8% versus 78,2%) a favor del grupo con HERCEPTIN®. Inmunogenicidad: en una paciente de las 903, se detectaron anticuerpos humanos antitrastuzumab, sin que tuviera reacciones alérgicas.
Contraindicaciones: HERCEPTIN® está contraindicado en pacientes con hipersensibilidad conocida al trastuzumab o a cualquier otro componente de la fórmula.
Precauciones generales: El tratamiento con HERCEPTIN® debe iniciarse siempre bajo la supervisión de un médico con experiencia en el tratamiento de pacientes con cáncer. Reacciones adversas graves descritas con poca frecuencia a la infusión de HERCEPTIN® son: disnea, hipotensión, sibilancias, broncoespasmo, taquicardia, reducción en la saturación de oxígeno y dificultad para respirar. La infusión con HERCEPTIN® debe ser interrumpida y la paciente vigilada hasta la resolución de cualquiera de los síntomas observados. Con medidas de apoyo como la administración de oxígeno, beta-agonistas o corticosteroides, se han tratado con éxito las reacciones graves (ver Reacciones secundarias y adversas). En casos muy raros, estas reacciones se han asociado a un curso clínico que culmine fatalmente. El riesgo de una reacción fatal por la infusión de HERCEPTIN® puede incrementarse en las pacientes que han experimentado disnea de reposo por las complicaciones propias de la enfermedad avanzada o a comorbilidades. Es por eso que este tipo de pacientes deben ser tratados con extrema precaución y se debe considerar el riesgo-beneficio de la terapia individualmente. Rara vez se han reportado efectos secundarios pulmonares severos con el uso de HERCEPTIN®. Estos eventos raros ocasionalmente han tenido un resultado fatal. Asimismo, se han reportado casos raros de infiltrados pulmonares, síndrome de insuficiencia respiratoria aguda, neumonía, neumonía intersticial, derrame pleural, edema pulmonar agudo e insuficiencia respiratoria. Estos eventos pueden presentarse como parte de una reacción relacionada a la infusión o con un inicio retrasado. Las pacientes con una neumonía intrínseca sintomática o con un extenso involucramiento tumoral de los pulmones, causante de disnea en reposo, pueden estar en mayor riesgo de reacciones severas (ver Reacciones secundarias y adversas). Se ha observado insuficiencia cardíaca (clase II-IV de la clasificación de la New York Heart Association [NYHA]) en las pacientes que recibieron terapia con HERCEPTIN® solo o en combinación con paclitaxel seguido de régimen quimioterapéutico que contiene antraciclinas (doxorrubicina o epirrubicina). Esta reacción puede ser de moderada a severa y se ha asociado a los casos de muerte (ver Reacciones secundarias y adversas). Deben extremarse las precauciones cuando se administre HERCEPTIN® a pacientes con insuficiencia cardíaca sintomática, antecedentes de hipertensión o con enfermedad coronaria preexistente, así como en el cáncer de mama temprano, de pacientes con una fracción de eyección ventricular izquierda (FEVI) del 55% o menor. Los candidatas al tratamiento con HERCEPTIN®, en especial las que ya han sido expuestas a antraciclinas y ciclofosfamida, deben someterse previamente a un reconocimiento médico para valorar su estado cardíaco, consistente en: anamnesis, exploración física, ECG, ecocardiografía y/o ventriculografía isotópica (MUGA, por sus siglas en inglés). Se debe evaluar de forma adecuada y cuidadosa el riesgo-beneficio que corre la paciente al utilizarse HERCEPTIN®. En CMT, los siguientes pacientes fueron excluidos del estudio HERA, por lo que no hay datos sobre el balance riesgo-beneficio, y consecuentemente, no se puede recomendar tratamiento en dichos pacientes: historia documentada de insuficiencia cardíaca congestiva; arritmias de alto riesgo no controladas; angina pectoris que requiera medicamento; enfermedad valvular clínicamente significativa; evidencia de infarto transmural en el ECG; hipertensión mal controlada. La función cardíaca debe ser vigilada a lo largo de todo el tratamiento (por ejemplo, cada tres meses). Este monitoreo puede ayudar a identificar a las pacientes que han desarrollado alguna disfunción cardíaca. Las pacientes que han desarrollado disfunción cardíaca asintomática deben ser monitoreadas con mayor frecuencia (por ejemplo, cada 6-8 semanas). Si las pacientes presentan un continuo decremento en la función ventricular izquierda, pero permanece asintomática, el médico puede considerar la interrupción de la terapia con HERCEPTIN® si es que no se han observado beneficios clínicos durante la terapia. Si la fracción de eyección ventricular izquierda (FEVI) cae en 10 puntos del valor basal o a menos del 50%, se debe detener la administración de HERCEPTIN® y repetir la determinación de la FEVI dentro de las 3 semanas siguientes, aproximadamente. Si entretanto la FEVI no ha mejorado o incluso ha disminuido aún más, se considerará decididamente la conveniencia de retirar HERCEPTIN®, salvo si se estima que los beneficios para la paciente sobrepasan a los riesgos. Si se desarrolla insuficiencia cardíaca sintomática durante el tratamiento con HERCEPTIN®, ésta deberá ser tratada con los medicamentos necesarios para este propósito. La interrupción de la terapia con HERCEPTIN® debe ser considerada de manera prioritaria en las pacientes en los que se desarrolla insuficiencia cardíaca significativa, a menos que los beneficios se estimen mayores a los riesgos. No se ha estudiado de forma prospectiva la seguridad toxicológica de continuar o reiniciar la administración de HERCEPTIN® para las pacientes que han experimentado cardiotoxicidad. Sin embargo, la mayoría de las pacientes que desarrollaron insuficiencia cardíaca durante los estudios clínicos han mejorado con tratamientos comunes, que incluyen diuréticos, glucósidos cardíacos y/o inhibidores de la enzima convertidora de la angiotensina. La mayoría de las pacientes con síntomas cardíacos y evidencia de beneficios clínicos por el tratamiento con HERCEPTIN® continuaron durante una terapia que consta de una dosis semanal sin que se presentaran eventos cardíacos adicionales. El uso de alcohol bencílico como conservador en el agua bacteriostática para inyectables en el frasco ámpula multidosis de 440mg ha sido asociado toxicidad en recién nacidos y niños menores de 3 años. Cuando se vaya a administrar HERCEPTIN® a una paciente con antecedentes de alergia al alcohol bencílico, se debe reconstituir el medicamento con agua para inyectables y utilizarse una dosis única de HERCEPTIN® por frasco ámpula; el resto debe desecharse.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: categoría B. No se sabe si HERCEPTIN® puede causar daño fetal si se administra a mujeres embarazadas o si puede afectar a la capacidad reproductiva. Dado que los estudios de reproducción animal no tienen siempre un valor pronóstico de la respuesta en el ser humano, debe evitarse el tratamiento con HERCEPTIN® durante el embarazo, salvo que los beneficios esperados para la madre sean mayores que los riesgos potenciales para el feto (ver Precauciones generales y Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad). Se desconoce si trastuzumab es secretado en la leche humana. Dado que la IgG humana se secreta en la leche, y desconociéndose el daño potencial para el niño, debe evitarse la lactancia durante el tratamiento con HERCEPTIN® (ver Farmacocinética).
Reacciones secundarias y adversas: Cáncer de mama metastásico (CMM): experiencia de los ensayos clínicos: en dos de los estudios clínicos iniciales en que las pacientes recibieron HERCEPTIN® como monoterapia o en combinación con paclitaxel, aproximadamente en 50% de ellos se esperó encontrar reacciones adversas. Las reacciones adversas más comunes fueron síntomas relacionados con la infusión, así como fiebre y escalofríos seguidos de la primera dosis de HERCEPTIN®. Las reacciones adversas atribuidas a HERCEPTIN® que se presentaron en ≥10% de las pacientes tratadas dentro de los 2 estudios clínicos iniciales fueron las siguientes: generales: dolor abdominal, astenia, dolor de pecho, escalofríos, fiebre, cefalea y dolor. Aparato digestivo: diarrea, náuseas y vómitos. Aparato locomotor: artralgia y mialgia. Piel y anexos: erupción cutánea. Las reacciones adversas atribuidas a HERCEPTIN® se presentaron en > 1% y < 10% de las pacientes tratadas dentro de los dos estudios clínicos iniciales fueron las siguientes: generales: dolor de espalda, enfermedad de tipo gripal, infección, dolor de cuello, malestar general y hipersensibilidad. Aparato cardiovascular: vasodilatación, taquiarritmia supraventricular, hipotensión, insuficiencia cardíaca, cardiomiopatía y palpitaciones. Aparato digestivo: anorexia, estreñimiento y dispepsia. Sistema hematopoyético: leucopenia. Metabolismo: edema periférico y edema. Aparato locomotor: dolor óseo. Sistema nervioso: ansiedad, depresión, mareo, insomnio, parestesias, somnolencia, hipertonía y neuropatía periférica. Aparato respiratorio: asma, aumento de la tos, disnea, epistaxis, alteración pulmonar, derrame pleural, faringitis, rinitis y sinusitis. Aparato urogenital: infección de vías urinarias. Piel y anexos: prurito, sudoración, trastornos de las uñas, piel seca, alopecia, acné y erupción maculopapular. En otro estudio clínico aleatorizado, se administró docetaxel, con o sin HERCEPTIN®, a pacientes con cáncer de mama metastásico.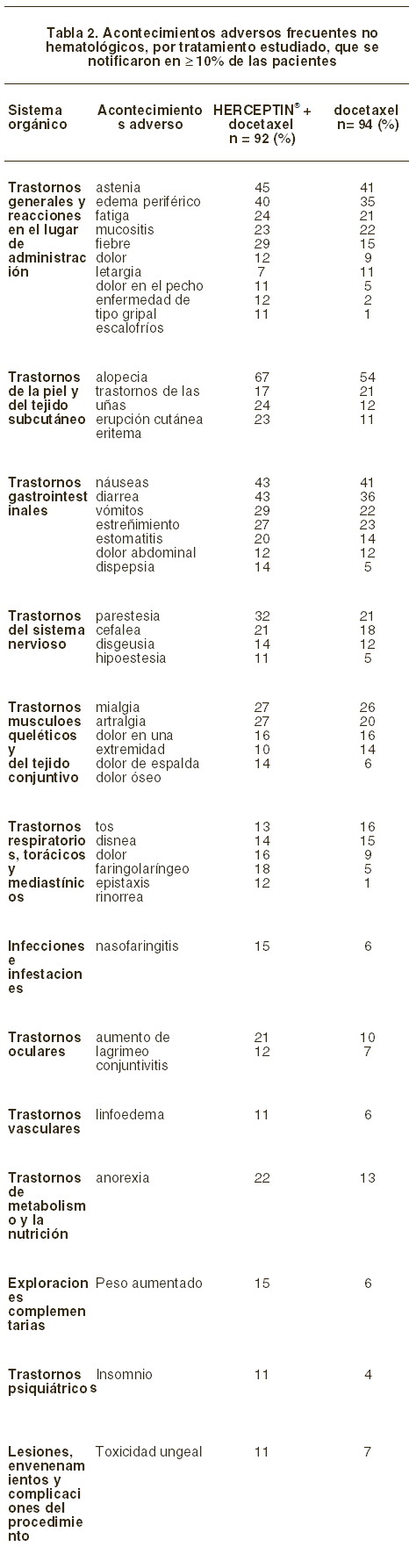
El proyecto TAnDEM es un estudio aleatorio, abierto, que comparó anastrozole solo contra anastrozole más HERCEPTIN® en pacientes con cáncer de mama metastásico (ver sección Estudios clínicos y de eficacia). En el estudio TAnDEM, no hubo cambio alguno en la naturaleza o frecuencia de EA, en comparación con estudios previos en población metastásica. Cáncer de mama temprano (CMT): el ensayo clínico HERA es un estudio abierto y aleatorizado, en pacientes con cáncer de mama temprano HER2 positivo (ver Farmacodinamia). La tabla 3 muestra los eventos adversos, por tratamiento estudiado, notificados en ≥1% de las pacientes al cabo de 1 año.
La información siguiente es de interés para todas las indicaciones: síntomas asociados a la infusión: durante la primera infusión con HERCEPTIN®, se observa habitualmente en las pacientes escalofríos y/o fiebre. Otros signos y/o síntomas pueden incluir náusea, vómito, dolor, escalofríos, cefalea, tos, mareos, erupción cutánea, astenia e hipertensión. Esos síntomas son generalmente de intensidad media a moderada y ocurren con poca frecuencia en las infusiones subsecuentes con HERCEPTIN®. Estos síntomas pueden ser tratados con analgésicos/antipiréticos, como por ejemplo meperidina o paracetamol, o un antihistamínico, como por ejemplo difenhidramina (ver Dosis y vía de administración). Algunas reacciones adversas a la infusión con HERCEPTIN® incluyen: disnea, hipotensión, sibilancias, broncoespasmo, taquicardia, reducción en la saturación de oxígeno e insuficiencia respiratoria que puede ser seria e incluso potencialmente fatal (ver Precauciones generales). Reacciones alérgicas: se han observado reacciones anafilácticas en casos aislados. Cardiotoxicidad: en pacientes tratadas con HERCEPTIN® se han observado signos y síntomas de disfunción cardíaca como disnea, ortopnea, incremento de la tos, edema pulmonar, ritmo de galope (S3) o reducción en la fracción de eyección (ver Precauciones generales). Dependiendo de los criterios utilizados para definir la disfunción cardíaca, la incidencia de los estudios pivote metastásicos varió entre el 9% y el 12% en el subgrupo tratado con HERCEPTIN® + paclitaxel, en comparación con el 1%-4% para el subgrupo que recibió paclitaxel solo. Para la monoterapia con HERCEPTIN®, la tasa fue del 6%-9%. La tasa mas elevada de disfunción cardíaca fue observada en las pacientes que estaban recibiendo HERCEPTIN® + antraciclina/ciclofosfamida (27%-28%), la cual fue significativamente mas elevada que la tasa reportada para las pacientes del subgrupo tratado con antraciclina/ciclofosfamida sola (7%-10%). En un estudio posterior con control prospectivo de la función cardíaca, la incidencia de insuficiencia cardíaca sintomática fue del 2,2% en las pacientes tratadas con HERCEPTIN® y docetaxel, comparado con el 0% en las que recibieron docetaxel solo. En el estudio HERA, se observó insuficiencia cardíaca de clase III-IV de la NYHA en el 0,6% de las pacientes del grupo de un año. Debido a que la vida media terminal de HERCEPTIN® es de 28,5 días (95% de intervalo de confianza, 25,5-32,8 días), el trastuzumab puede persistir en la circulación hasta por 20 semanas (95% de intervalo de confianza, 18-24 semanas) después de suspender el tratamiento. Debido a que el uso de antraciclina durante este período podría posiblemente asociarse con un incremento en el riesgo de disfunción cardíaca, se recomienda una evaluación completa de los riesgos, versus los beneficios potenciales, además de un cuidadoso monitoreo cardíaco. Toxicidad hematológica: la toxicidad hematológica es infrecuente tras la administración de HERCEPTIN® como monoterapia a pacientes con enfermedad metastásica. Se han observado leucopenia, trombocitopenia y anemia de grado 3 según la clasificación de la OMS, en < 1% de las pacientes. No se ha observado toxicidad de grado 4 de la clasificación de OMS. Hubo un incremento en la toxicidad hematológica grado 3 y 4 de la clasificación de la OMS en pacientes tratados con la combinación HERCEPTIN® más paclitaxel, con respecto a las pacientes que estaban recibiendo paclitaxel solo (34% vs. 21%). La toxicidad hematológica también aumentó en las pacientes que recibieron HERCEPTIN® y docetaxel en comparación con las tratadas con docetaxel solo (32% vs. 22% con neutropenia de grado 3-4, según los criterios comunes de toxicidad del NCI-CTC). La incidencia de neutropenia febril/sepsis neutropénica también creció en las pacientes tratadas con HERCEPTIN® y docetaxel (23% vs. 17% en las que recibieron docetaxel solo). De acuerdo con los criterios NCI-CTC, en el estudio HERA hubo un 0,4% de pacientes tratadas con HERCEPTIN® que experimentaron un cambio de 3 o 4 grados del valor basal, frente al 0,6% en el grupo de observación. Toxicidad hepática y renal: se ha observado toxicidad hepática de grado 3 o 4 de la clasificación de la OMS en el 12% de las pacientes tras la administración de HERCEPTIN® como monoterapéutico contra la enfermedad metastásica. Esta toxicidad fue asociada con la progresión de la enfermedad en el hígado en el 60% de estas pacientes. La toxicidad hepática de grado 3 o 4 de la clasificación de OMS fue observada menos frecuentemente en las pacientes que recibieron HERCEPTIN® y paclitaxel en comparación con las pacientes que recibieron paclitaxel solo (7% vs. 15%). No se ha detectado toxicidad renal de grado 3 o 4 de la clasificación de la OMS. Diarrea: de las pacientes tratadas con monoterapia contra la enfermedad metastásica de HERCEPTIN® el 27% experimentaron diarrea. Además se observó un incremento en la incidencia de diarrea de media a moderada intensidad, en las pacientes que recibieron HERCEPTIN® en combinación con paclitaxel con respecto a las pacientes que recibieron paclitaxel solo. En el estudio HERA, el 7% de las pacientes tratadas con HERCEPTIN® presentaron diarrea. Infección: se ha registrado un incremento en la incidencia de infecciones, principalmente infecciones leves de las vías respiratorias altas de poca importancia clínica o infecciones por catéter, sobre todo entre las pacientes tratados con HERCEPTIN® en asociación con paclitaxel, en relación con las que recibieron paclitaxel solo. Reacciones adversas serias: cuando menos un caso de las siguientes reacciones adversas ocurrieron en cuando menos uno de las pacientes tratadas con HERCEPTIN® solo o en combinación con paclitaxel en los estudios clínicos: generales: reacción alérgica, anafilaxis y shock anafiláctico, ataxia, sepsis, fiebre, astenia, escalofríos, cefalea, parestesias, dolor en el pecho y fatiga. Aparato cardiovascular: cardiomiopatía, insuficiencia cardíaca congestiva e incremento en disminución en la fracción de eyección, hipotensión, derrame pericárdico, bradicardia y enfermedad cerebrovascular. Aparato digestivo: hepatitis, diarrea, náuseas y vómitos. Sistemas hematopoyético: leucemia, neutropenia febril, neutropenia y trombocitopenia. Infecciones: celulitis, erisipela. Aparato respiratorio: broncoespasmo, insuficiencia respiratoria y edema agudo pulmonar. Piel y anexos: erupción cutánea. Experiencia postcomercialización: las siguientes reacciones adversas atribuidas a HERCEPTIN® se reportaron en por lo menos una paciente durante la experiencia postcomercialización: generales: síntomas relacionados con la infusión, edema periférico, dolor óseo, coma, meningitis, edema cerebral y pensamiento anormal. Aparato cardiovascular: insuficiencia cardíaca, shock cardiogénico, pericarditis e hipertensión. Aparato digestivo: pancreatitis, insuficiencia hepática e ictericia. Sistema hematopoyético: anemia y disminución de protrombina. Aparato locomotor: mialgia. Aparato respiratorio: disnea, hipoxia, edema laríngeo, insuficiencia respiratoria aguda, síndrome de sufrimiento respiratorio agudo, derrame pleural e infiltraciones pulmonares, neumonía, neumonitis y fibrosis pulmonar. Aparato urinario: glomerulonefropatía, insuficiencia renal. Piel y anexos: dermatitis y urticaria. Organos de los sentidos: pérdida de la audición.
Interacciones medicamentosas y de otro género: No se han realizado estudios formales de interacciones medicamentosas con HERCEPTIN® en el ser humano. No se han observado interacciones clínicamente significativas con la medicación concomitante utilizada en los ensayos clínicos (ver Farmacocinética).
Alteraciones en los resultados de pruebas de laboratorio: Se desconocen hasta la fecha.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Se han llevado a cabo estudios sobre la reproducción en macacos de Java, con dosis de HERCEPTIN® hasta 25 veces superiores a la dosis semanal de mantenimiento en el ser humano, de 2 mg/kg, sin que se evidenciara ninguna alteración en la fertilidad ni daño fetal. Sin embargo, al evaluar el riesgo de toxicidad reproductiva en humanos, es importante considerar el significado de la forma del receptor HER2 de los roedores en el desarrollo embrionario normal, así como la muerte embrionaria en ratones mutantes sin este receptor. Se observó transferencia placentaria de trastuzumab durante la fase temprano (días 20-50 de la gestación) y tardía (días 120-150 de la gestación) en el período de desarrollo fetal. Otros efectos (inmunogenecidad, prolongación del intervalo QT, etc.): en un estudio con monos Cynomolgus en período de lactancia, con dosis de HERCEPTIN® 25 veces superiores a las de mantenimiento semanal en el ser humano que es de 2 mg/kg, se observó que trastuzumab es secretado en la leche. La presencia de trastuzumab en el suero de monos lactantes no se ha asociado con ninguna reacción adversa sobre su crecimiento o desarrollo desde el nacimiento hasta el primer mes de edad.
Dosis y vía de administración: Manejo y eliminación: se deben emplear métodos asépticos adecuados. El frasco ámpula de 440 mg de HERCEPTIN® se reconstituye con 20 ml de agua bacteriostática para inyectables que contenga 1,1% de alcohol bencílico, como la que se suministra con el producto; de esta reconstitución se obtiene una solución multidosis, que contiene 21 mg/ml de trastuzumab, con un pH de aproximadamente 6,0. Se debe evitar el uso de otras soluciones como reconstituyentes. El HERCEPTIN® debe ser manejado cuidadosamente durante su reconstitución. La formación de espuma excesiva durante el proceso de reconstitución o de agitación puede provocar problemas con la cantidad de HERCEPTIN® que se retira del frasco ámpula. Instrucciones para la reconstitución para el vial de 440 mg: utilizando una jeringa estéril, lentamente inyecte los 20 ml del agua bacteriostática para inyectables al vial que contiene el liofilizado de HERCEPTIN®, colocando la aguja dentro del polvo liofilizado. Mueva el vial con un suave movimiento rotatorio para ayudar a la reconstitución. No agite. Es común que se forme una delgada capa de espuma después de la reconstitución del producto. Mantenga el vial sin movimiento durante aproximadamente 5 minutos. La preparación reconstituida de HERCEPTIN® es una solución transparente, incolora a amarillo claro y no debe contener partículas visibles. Instrucciones para la dilución: el volumen de solución requerida se calcula tomando en cuenta la dosis de carga de trastuzumab de 4 mg/kg y las dosis de mantenimiento de 2 mg/kg:
El volumen de solución requerida se calcula tomando en cuenta la dosis de carga de trastuzumab de 8 mg/kg y las dosis de mantenimiento de 6 mg/kg:
Una vez realizado el cálculo, la cantidad resultante de solución se extrae del vial y se añade a una bolsa de infusión con 250 ml de solución de cloruro de sodio al 0,9%. No se debe utilizar solución glucosada (5%) (ver Incompatibilidades). La bolsa debe invertirse cuidadosamente varias veces para mezclar la solución y evitar la formación de espuma. Antes de su administración, los medicamentos parenterales deben inspeccionarse visualmente para averiguar si contienen partículas o si están decolorados. Una vez preparada, la infusión debe administrarse inmediatamente (ver Recomendaciones para almacenamiento). Incompatibilidades: no se ha observado que existan incompatibilidades entre el HERCEPTIN® y las bolsas de cloruro de polivinilo, polietileno o polipropileno. La solución de dextrosa al 5% no debe ser utilizada porque causa la agregación de la proteína. HERCEPTIN® no debe ser mezclado o diluido con otros medicamentos. Dosificación: la prueba positiva para HER2 es mandatoria para el comienzo de la terapia con HERCEPTIN®. El HERCEPTIN® debe administrarse en infusión intravenosa. El HERCEPTIN® no debe administrarse en inyección intravenosa rápida o en bolo IV. Dosis semanal: dosis de carga: se recomienda administrar una dosis inicial de HERCEPTIN® de 4 mg/kg en infusión intravenosa de 90 minutos. Es preciso observar a las pacientes por si llegaran a presentar fiebre, escalofríos u otros síntomas relacionados con la infusión (ver Reacciones secundarias y adversas), en cuyo caso debe interrumpirse la infusión intravenosa, para reanudarla una vez desaparecidos los síntomas. Dosis mantenimiento: para el tratamiento de mantenimiento se recomienda una dosis semanal de HERCEPTIN® de 2 mg/kg. Si la dosis previa se toleró bien, las dosis de mantenimiento pueden administrarse en infusión intravenosa de 30 minutos. Es preciso observar a las pacientes por si llegaran a presentar fiebre, escalofríos u otros síntomas relacionados con la infusión (ver Reacciones secundarias y adversas). En los estudios clínicos, HERCEPTIN® se administró hasta la progresión de la enfermedad. Dosis alternativa cada tres semanas: dosis de carga (inicial) de 8 mg/kg al cabo de 3 semanas, seguida de 6 mg/kg al cabo de 3 semanas, y a continuación 6 mg/kg cada 3 semanas, en infusión de aproximadamente 90 minutos. Las pacientes con cáncer de mama temprano deben recibir tratamiento durante 1 año o hasta la recurrencia de la enfermedad. Si una paciente se salta una dosis de trastuzumab en una semana o menos, debe recibir después la dosis habitual (6 mg/kg) lo antes posible (sin esperar hasta el próximo ciclo programado). Las dosis subsecuentes de mantenimiento de 6 mg/kg deben administrarse cada 3 semanas, según el plan anterior. Si una paciente se salta una dosis de trastuzumab en más de una semana, debe recibir una nueva dosis de carga (8 mg/kg en aproximadamente 90 minutos). Las dosis subsecuentes de mantenimiento de 6 mg/kg deben administrarse cada 3 semanas, contadas desde ese momento. Reducción de la dosis: durante los ensayos clínicos, no se efectuó ninguna reducción de la dosis de HERCEPTIN®. Las pacientes pueden continuar el tratamiento con HERCEPTIN® durante los períodos de mielodepresión reversible inducida por la quimioterapia, pero se les debe vigilar estrechamente para detectar posibles complicaciones de una neutropenia durante este período. Deben seguirse las instrucciones específicas para reducir o mantener la dosis de quimioterapia. Pautas posológicas especiales: ancianas: los datos indican que el comportamiento farmacocinético de HERCEPTIN® no cambia con la edad (ver Farmacocinética en poblaciones especiales). En los ensayos clínicos, las pacientes ancianas no recibieron dosis reducidas de HERCEPTIN®. Niñas: no se ha estudiado la seguridad toxicológica y la eficacia de HERCEPTIN® en pacientes pediátricas.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: No se conoce ningún caso de sobredosis durante los ensayos clínicos. Dosis superiores a 10 mg/kg no han sido probadas.
Presentación(es): Caja con un frasco ámpula con 440 mg y un vial con 20 ml de agua bacteriostática para inyectables con instructivo anexo.
Recomendaciones sobre almacenamiento: Frascos ámpula: consérvese en refrigeración, de 2 a 8°C. Período de validez de la solución reconstituida: frasco ámpula de 440 mg: la solución reconstituida con agua bacteriostática para inyectables, para los viales de 440 mg de HERCEPTIN®, como la que se incluye en el producto, se mantiene estable durante 28 días si se conserva en refrigeración de 2°C a 8°C. La solución reconstituida contiene un conservador y, por consiguiente, es idónea para su uso como multidosis. El resto de la solución reconstituida debe desecharse al cabo de 28 días. Si se usa agua estéril para reconstituir los viales de 440 mg, la solución es estable sólo por 24 horas, y debe ser desechado el sobrante. Desde el punto de vista microbiológico, la solución para infusión de HERCEPTIN® debe ser utilizada inmediatamente. Si no se diluye inmediatamente, la duración y las condiciones de conservación del producto antes de su disolución son responsabilidad del usuario. Habitualmente, no debería sobrepasar las 24 horas a 2°C a 8°C, salvo que la dilución se haya llevado a cabo bajo condiciones asépticas controladas y validadas. Período de estabilidad de la solución para infusión con el producto reconstituido: una vez diluida la solución de HERCEPTIN® en solución de cloruro de sodio al 0,9% dentro de las bolsas de cloruro de polivinilo o polietileno, es estable durante 24 horas si se conserva en refrigeración, de 2°C a 8°C. Se ha demostrado que las diluciones de HERCEPTIN® permanecen estables hasta por 24 horas (no debe conservarse a más de 30°C). Sin embargo, si la solución de HERCEPTIN® no contiene un conservador efectivo, las soluciones reconstituida y diluida deben conservarse en refrigeración, de 2°C a 8°C. Desde el punto de vista microbiológico, la solución para infusión de HERCEPTIN® debe ser utilizada inmediatamente. Si no se utiliza inmediatamente, la duración y las condiciones de conservación del producto antes de su utilización son de la responsabilidad del usuario. Habitualmente, no debería sobrepasar las 24 horas a 2°C a 8°C, salvo que la dilución se haya llevado a cabo bajo condiciones asépticas controladas y validadas. El HERCEPTIN® no deberá utilizarse después de la fecha de caducidad, indica con EXP en el envase.
Leyendas de protección: Su venta requiere receta médica. No se deje al alcance de los niños. Literatura exclusiva para médicos. No se use en el embarazo y la lactancia.
Nombre y domicilio del laboratorio: HERCEPTIN® Vial multidosis de