INCRESINA®
TAKEDA
Denominación genérica: Alogliptina.
Forma farmacéutica y formulación: Tableta. Cada tableta contiene: Benzoato de Alogliptina. Equivalente a 6.25mg, 12.5mg y 25mg de alogliptina.
Indicaciones terapéuticas: INCRESINA® es un inhibidor de la dipeptidil-peptidasa-4 (DPP-4), indicada como un complemento a la dieta y al ejercicio para mejorar el control glicémico en adultos con diabetes mellitus tipo 2 en las siguientes condiciones clínicas: Como monoterapia en pacientes con un control glicemico inadecuado con dieta y ejercicio. Como un complemento al tratamiento con metformina. Como un complemento al tratamiento con una tiazolidinediona (TZD). Como un complemento al tratamiento con una sulfonilurea (SU). Como un complemento al tratamiento con insulina.
Farmacocinética y farmacodinamia: Las tabletas de INCRESINA® contienen como ingrediente activo alogliptina, el cual es un inhibidor selectivo y potente de la actividad enzimática de la DPP-4. Las concentraciones de hormonas incretinas como el péptido 1 similar al glucagón (GLP-1) y el péptido insulinotrópico glucosa-dependiente (GIP) se liberan al torrente sanguíneo a partir del intestino pequeño en respuesta a los alimentos. Estas hormonas causan la liberación de insulina de las células beta pancreáticas de manera glucosa-dependiente, pero son desactivadas por la enzima DPP-4 en cuestión de minutos. El GLP-1 también disminuye la secreción de glucagón en las células beta pancreáticas, reduciendo la producción de glucosa hepática. En pacientes con diabetes tipo 2, las concentraciones de GLP-1 disminuyen pero se mantiene la respuesta de insulina al GLP-1. La alogliptina es un inhibidor sumamente potente y selectivo de dipeptidil peptidasa 4 (DPP-4) que reduce la desactivación de hormonas incretinas, aumentando así sus concentraciones y reduciendo las concentraciones durante el ayuno y las posprandiales de manera glucosa-dependiente en pacientes con diabetes mellitus tipo 2. Alogliptina se une selectivamente e inhibe la DPP-4 pero no la actividad de DPP-8 o DPP-9 in vitro en concentraciones que se aproximan a las exposiciones terapéuticas. Farmacocinética: Absorción: La farmacocinética de INCRESINA® ha sido estudiada en sujetos sanos y pacientes con diabetes tipo 2. Después de la administración de dosis orales, simples de hasta 800 mg en sujetos sanos, la alogliptina fue rápidamente absorbida, alcanzando concentraciones plasmáticas máximas (Tmax mediana) entre 1 y 2 horas después de la administración. A la dosis de 25 mg, INCRESINA® fue eliminada de una manera bifásica, con una vida media terminal (T1/2) de aproximadamente 21 horas. Después de la administración de dosis múltiples de hasta 400 mg por 14 días en pacientes con diabetes tipo 2, la acumulación de alogliptina fue mínima con un aumento en las exposiciones totales (es decir, AUC) y máximas (es decir, Cmax) de alogliptina de 34% y 9%, respectivamente. Las exposiciones totales y máximas a la alogliptina aumentaron de manera proporcional a través de las dosis únicas y las dosis múltiples de alogliptina que iban de 25 mg a 400 mg. El coeficiente inter-sujeto de variación para la AUC de alogliptina fue bajo (17%). La farmacocinética de INCRESINA® ha demostrado ser similar en sujetos sanos y en pacientes con diabetes tipo 2. La biodisponibilidad absoluta de la alogliptina es de aproximadamente 100%. La administración de INCRESINA® con alimentos ricos en grasas no da como resultado cambios significativos en la exposición total y máxima a la alogliptina. Por lo tanto, INCRESINA® se puede administrar con o sin alimentos. Distribución: Después de una infusión intravenosa única de 12.5 mg de alogliptina en sujetos sanos, el volumen de distribución durante la fase terminal fue de 417L, lo que indica que el fármaco está bien distribuido en los tejidos. La alogliptina se une 20% a las proteínas plasmáticas. Después de una dosis única por vía oral de 25 mg de alogliptina en sujetos sanos, el volumen aparente de distribución durante la fase terminal fue de 609.61 litros, lo que indica que el fármaco está bien distribuido en los tejidos. Metabolismo: La alogliptina no se metaboliza de forma extensiva y el 60% al 71% de la dosis se excreta en forma de fármaco inalterado en la orina. Se detectaron dos metabolitos menores tras la administración de una dosis oral de [14C] alogliptina, alogliptina N-desmetilada, M-I ( < 1% del compuesto original), y alogliptina N-acetilada, M-II ( < 6% del compuesto original). M-I es un metabolito activo y un inhibidor de la DPP-4 similar a la molécula original; M-II no muestra ninguna actividad inhibidora con respecto a la DPP-4 ni otras enzimas relacionadas con la DPP. Los datos in vitro indican que CYP2D6 y CYP3A4 contribuyen al metabolismo limitado de alogliptina. La alogliptina existe predominantemente en forma de (R)-enantiómero ( > 99%) y sufre una conversión quiral a (S)-enantiómero escasa o nula in vivo. El (S)-enantiómero es indetectable en la dosis de 25 mg. Excreción: La vía de eliminación primaria de la radioactividad derivada de [14C] alogliptina se hizo por excreción renal (76%) y 13% se recuperó en las heces, por lo que se alcanzó una recuperación total de 89% de la dosis radioactiva administrada. La depuración renal promedio de alogliptina (9.6 L/hr) indica que alguna secreción tubular renal activa y depuración sistémica fue de 14.0 L/hr. Farmacodinamia: La administración de una dosis única de INCRESINA® a individuos sanos dio como resultado una inhibición máxima de la DPP-4 en 2-3 horas después de la dosificación. El pico de inhibición de la DPP-4 excedió 93% de las dosis de 12.5 mg a 800 mg. La inhibición de la DPP-4 permaneció por encima de 80% a las 24 horas en las dosis mayores o iguales a 25 mg. La exposición pico y total de más de 24 horas para GLP-1 activo fue de tres a cuatro veces mayor con INCRESINA® (en dosis de 25 a 200 mg) que con placebo. En un estudio doble ciego, controlado con placebo de 16 semanas con INCRESINA® de 25 mg, se demostró que ésta disminuye el glucagón postprandial mientras que incrementa los niveles postprandiales de GLP-1 activo, en comparación con el placebo durante un periodo de ocho horas después de una comida estándar. Aún no está claro por qué estos resultados se relacionan con cambios en el control glicemico general de los pacientes con diabetes mellitus tipo 2. En este estudio, se demostró que INCRESINA® de 25 mg disminuye en dos horas la glucosa postprandial en comparación con el placebo (-30 mg/dL vs 17 mg/dL, respectivamente). La administración de dosis múltiples de alogliptina en pacientes con diabetes mellitus tipo 2 dio como resultado una inhibición máxima de la DPP-4 en una a dos horas y excedió 93% en todas las dosis (25, 100 y 400 mg) después de una dosis única y después de 14 días de una sola dosis diariamente. En estas dosis de NESINA®, la inhibición de la DPP-4 permaneció por encima de 81% a las 24 horas y después de 14 días de dosificación diaria.
Contraindicaciones: Este producto está contraindicado en pacientes con hipersensibilidad conocida a la alogliptina o a cualquiera de sus componentes. Menores de 18 años.
Precauciones generales: Pancreatitis aguda: Se han reportado eventos de pancreatitis aguda posteriores a la comercialización de la alogliptina y estos han sido relacionados con otros inhibidores de DPP-4. Después de iniciar la toma de alogliptina, los pacientes deben estar en observación para determinar la presencia de signos y síntomas de pancreatitis. Si existe la sospecha de pancreatitis, la alogliptina debe suspenderse de inmediato y debe iniciarse el manejo correspondiente. Reacciones de hipersensibilidad: Se han reportado eventos de reacciones serias de hipersensibilidad posteriores a la comercialización en pacientes tratados con alogliptina como angioedema y reacciones adversas severas de tipo cutáneo, incluyendo el síndrome de Stevens-Johnson, los cuales se han asociado con otros inhibidores de DPP-4. En caso de sospechar una reacción de hipersensibilidad, debe suspenderse la alogliptina. Efectos hepáticos: Se han recibido reportes de disfunción hepática, incluyendo insuficiencia renal, posteriores a la comercialización. Los pacientes deben estar en observación en caso de presentarse posibles anormalidades hepáticas. Deben hacerse pruebas hepáticas en pacientes que presenten síntomas que indiquen daño hepático. De encontrarse alguna anormalidad sin poder establecer una etiología alternativa, debe considerarse la suspensión de alogliptina. Hipoglucemia: Es sabido que la insulina y los secretagogos de insulina, como las sulfonilureas, causan hipoglucemia. Por lo tanto, puede ser necesaria una dosis menor de insulina o de secretagogo de insulina para minimizar el riesgo de hipoglucemia al usarse en combinación con la alogliptina.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: Categoría B de Embarazo: Este fármaco puede usarse durante el embarazo sólo si los beneficios son mayores que los riesgos para la madre y el feto. Se han realizado estudios de desarrollo y reproducción en ratas y conejos con dosis que produjeron exposiciones de aproximadamente 180 y 149 veces, respectivamente, el promedio de exposición humana con la dosis clínica recomendada, y no indicaron que hubiera evidencia de discapacidad en fertilidad y/o daño al feto a causa de la alogliptina. En las ratas preñadas se observó paso de barrera placentaria de la alogliptina. Lactancia: Se desconoce si la alogliptina se excreta en la lecha materna. Deben tomarse precauciones al administrar este fármaco durante la lactancia. La alogliptina se secreta en la leche de ratas lactantes en una proporción de 2:1 al plasma. Se desconoce si la alogliptina se excreta en la lecha humana. No se realizaron estudios con alogliptina en mujeres embarazadas.
Reacciones secundarias y adversas: Las reacciones adversas se enlistan por grupo sistémico y frecuencia. Las frecuencias se definen como muy común (≥ 1/10); común ((≥ 1/100 a < 1/10); poco común (≥ 1/1,000 a < 1/100); rara (≥ 1/10,000 a < 1/1000), muy rara ( < 1/10,000); desconocidas (no se pueden estimar con datos disponibles).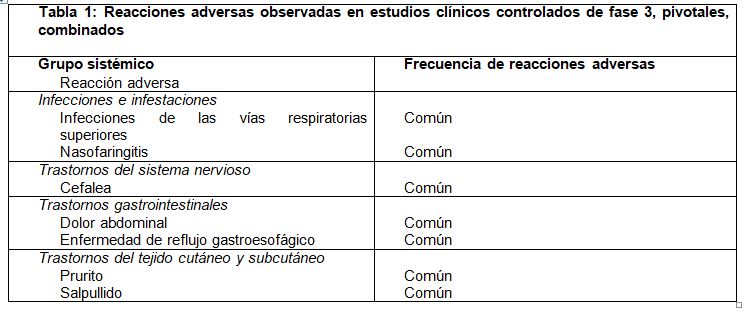
Experiencia poscomercial: La Tabla 2 muestra reacciones adversas adicionales que se han reportado de forma espontánea tras la comercialización del medicamento.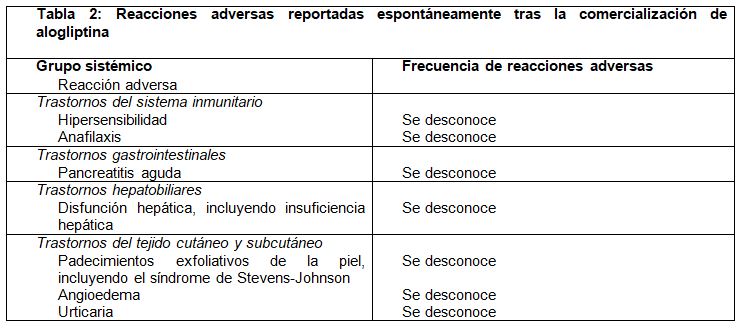
Interacciones medicamentosas y de otro genero: Evaluación in vitro de las interacciones medicamentosas. Los estudios in vitro indican que la alogliptina no es un inductor de las CYP1A2, CYP2B6, CYP2C9, CYP2C19 y CYP3A4, ni tampoco un inhibidor de las CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP3A4 y CYP2D6 en concentraciones clínicamente relevantes. Evaluación in vivo de las interacciones medicamentosas. Efectos de la alogliptina en la farmacocinética de otros fármacos. En los estudios clínicos, la alogliptina no aumentó de forma significativa la exposición sistémica a los siguientes fármacos que se metabolizan por isoenzimas CYP o que se excretan sin modificarse en la orina (Figura 1). No se recomienda ajustar las dosis de INCRESINA® en base a los resultados de los estudios farmacocinéticos descritos.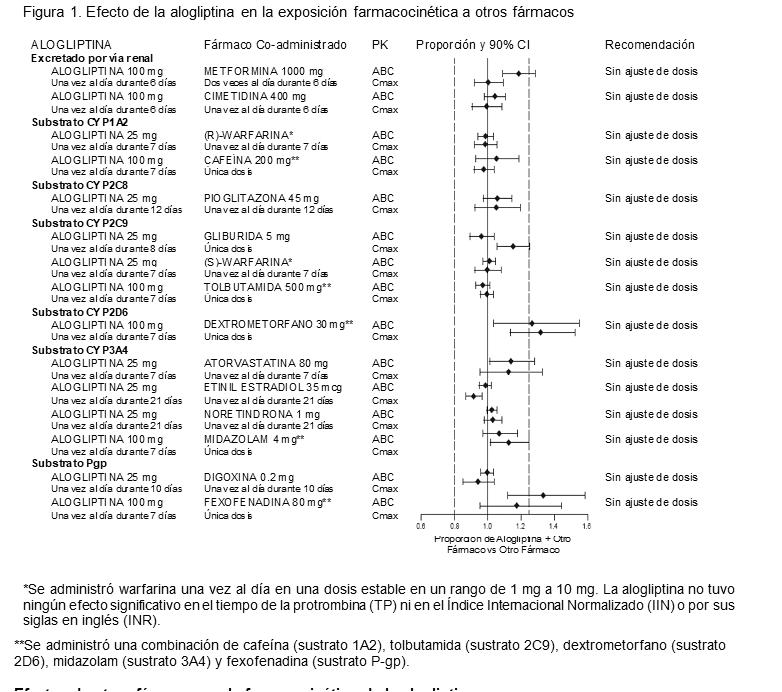
Efectos de otros fármacos en la farmacocinética de la alogliptina: No se observó ningún cambio significativo en la farmacocinética de la alogliptina cuando se administró INCRESINA® en combinación con los siguientes fármacos (Figura 2).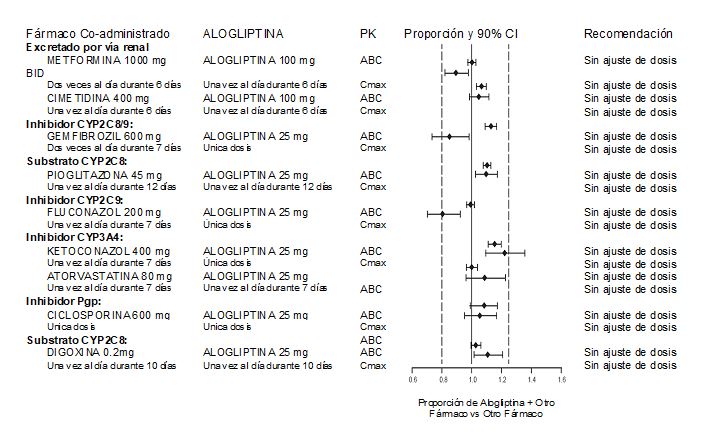
Alteraciones en los resultados de pruebas de laboratorio: No se observaron cambios clínicamente en la biometría hemática, química sérica y análisis de orina en pacientes tratados con INCRESINA®.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: La alogliptina no resultó carcinógena en estudios de carcinogénesis de 2 años realizados en ratas y ratones. En los estudios de carcinogénesis de 2 años, se administraron dosis orales de 50, 150 o 300 mg/kg/día y 75, 400, o 800 mg/kg/día en ratones y ratas, respectivamente. Las dosis máximas empleadas en los estudios con ratones y ratas (300 mg/kg/día y 800 mg/kg/día, respectivamente) proporcionaron márgenes de exposición que fueron de aproximadamente 51 y 399 veces, respectivamente, más altas que la AUC (0-24) en la dosis clínica de 25 mg. No se observaron tumores relacionados con el fármaco en las ratas en que se administraron dosis de hasta 75 mg/kg o aproximadamente 32 veces la máxima dosis clínica recomendada de 25 mg, con base en la exposición AUC. En dosis más altas (aproximadamente 308 veces la máxima dosis clínica recomendada de 25 mg), una combinación de adenomas y carcinomas tiroideos de células C aumentó en las ratas macho pero no en las hembras. No se observaron tumores relacionados con el fármaco en los ratones tras la administración de 50, 150 o 300 mg/kg de alogliptina por 2 años o hasta aproximadamente 51 veces la máxima dosis clínica recomendada de 25 mg, con base en una exposición AUC. La alogliptina no resultó mutagénica o clastogénica, con o sin activación metabólica, en la prueba Ames con S. typhimurium y E. coli o el ensayo citogenético en células del linfoma de ratones. La alogliptina resultó negativa en el estudio de micronúcleo en ratones en vivo. En un estudio de fertilidad en ratas, la alogliptina no tuvo efectos adversos en el desarrollo embrionario temprano, apareamiento o fertilidad en dosis de hasta 500 mg/kg, o aproximadamente 172 veces la dosis clínica basada en la exposición del medicamento en plasma (AUC). Toxicología animal y/o farmacología: La administración de dosis de hasta 400 mg/kg/día en ratas por 26 semanas o 200 mg/kg/día en perros por 39 semanas no dio como resultado ningún efecto adverso toxicológico. Estas dosis administradas en ratas y perros proporcionaron múltiples exposiciones de aproximadamente 147 y 227 veces, respectivamente, la dosis clínica basada en la exposición al medicamento en plasma (AUC). La administración de alogliptina no provocó ningún tipo de lesión cutánea relacionada con el fármaco en changos, un hallazgo que se ha venido observando en estudios realizados con otros inhibidores de DPP-4. Estudios clínicos: INCRESINA® ha sido estudiado como monoterapia y en combinación con metformina, una sulfonilurea, una tiazolidindiona (ya sea sola o en combinación con metformina o una sulfonilurea), o insulina (ya sea sola o en combinación con metformina). Un total de 2239 pacientes con diabetes tipo 2 fueron repartidos aleatoriamente en 5 estudios de eficacia y seguridad clínica controlados por placebo, en doble ciego, conducidos para evaluar los efectos de la alogliptina sobre el control glucémico. En pacientes con diabetes tipo 2, el tratamiento con INCRESINA® produjo disminuciones clínicamente significativas en HbA1c en comparación al placebo en la Semana 26 (Figura 1).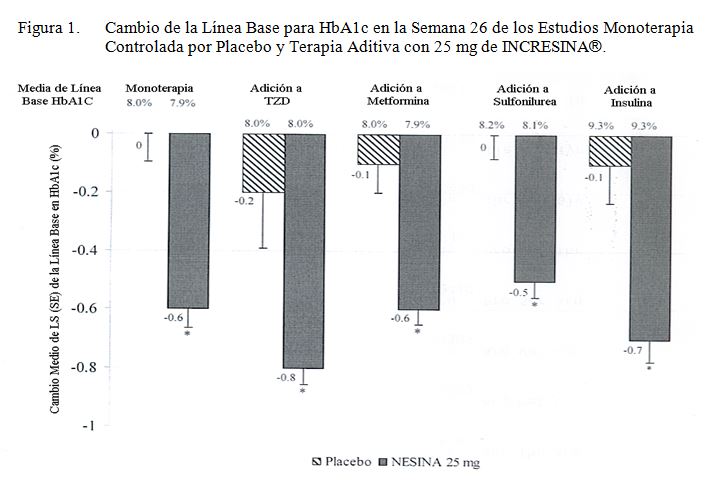
Monoterapia: Un estudio controlado por placebo de 26 semanas fue conducido para evaluar la eficacia y seguridad de la alogliptina como monoterapia. Un total de 329 pacientes controlados inadecuadamente con respecto a la dieta y al ejercicio solos (HbA1c media de línea base = 8.0%) dentro de 3 meses de la selección fueron repartidos aleatoriamente para recibir 12.5 mg de INCRESINA®, 25 mg de INCRESINA®, o placebo. Todos los pacientes entraron a un periodo de corrida de placebo, en ciego simple, de 4 semanas, antes de la repartición aleatoria. Después de la repartición aleatoria, todos los pacientes continuaron recibiendo instrucciones sobre la dieta y el ejercicio. Los pacientes quienes fallaron en cumplir las metas glucémicas definidas por los estudios específicos, fueron rescatados y se les permitió entrar a un estudio de extensión con rótulos a la vista con INCRESINA®. Tratamiento con 25 mg de INCRESINA® una vez al día dio como resultado mejoramientos significativos de la línea base en HbA1c y FPG en comparación al placebo a la Semana 26 (Tabla 3). También, significativamente, menos pacientes que recibieron 25 mg de INCRESINA® (8%) requerido en rescate hiperglucémico en comparación con aquellos que recibieron placebo (30%) durante el estudio. El mejoramiento en HbA1c no fue afectado por el género, la edad o la raza. La etnicidad hispana o BMI de línea base. No obstante, los pacientes quienes entraron en el estudio a un nivel de HbA1c de línea base más alto, en general lograron un efecto de tratamiento mayor. Con base en un análisis pre-especificado por HbA1c de línea base, los pacientes quienes entraron al estudio con una HbA1c ≥8%, lograron una reducción media significativa de la línea base de -0.9% sobre INCRESINA® a 25 mg versus -0.2% con placebo a la Semana 26. No existió diferencia significativa entre INCRESINA® y el placebo en el cambio de cuerpo corporal a la Semana 26. Los efectos de los lípidos fueron también en general neutros.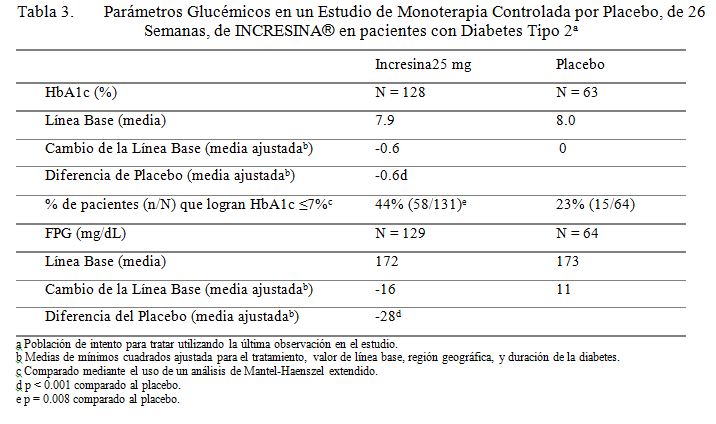
Terapia en Combinación: Terapia Aditiva a una Tiazolidindiona: Un estudio controlado por placebo de 26 semanas, fue conducido para evaluar la eficacia y la seguridad de INCRESINA® como terapia aditiva a la pioglitazona en pacientes con diabetes tipo 2. Un total de 493 pacientes controlados inadecuadamente en una tiazolidindiona sola o en combinación con metformina o una sulfonilurea (HbA1c media de línea base = 8.0%) fueron repartidos aleatoriamente para recibir 12.5 mg de INCRESINA®, 25 mg de INCRESINA®, o placebo. Los pacientes fueron mantenidos en una dosis estable de pioglitazona (dosis media = 35 mg) durante el periodo de tratamiento; aquellos quienes fueron también previamente tratados con metformina o sulfonilurea antes de la repartición aleatoria, fueron mantenidos en la terapia de combinación durante el periodo de tratamiento. Todos los pacientes entraron a un periodo de corrida con placebo, de ciego simple, de 4 semanas, antes de la repartición aleatoria. Después de la repartición aleatoria, todos los pacientes continuaron recibiendo instrucciones sobre la dieta y el ejercicio. Los pacientes quienes fallaron en cumplir las metas glucémicas definidas por el estudio, específicas, fueron rescatados y se les permitió entrar a un estudio de extensión con rótulos a la vista, con INCRESINA®. La adición de 25 mg de INCRESINA® una vez al día a la terapia con pioglitazona dio como resultado mejoramiento significativo de la línea base en HbA1c y FPG a la Semana 26, cuando se comparó a la adición del placebo (Tabla 4). Durante el estudio, 9% de los pacientes quienes estaban recibiendo 25 mg de INCRESINA requirieron rescate hiperglucémico en comparación con 12% de los pacientes que recibieron placebo. El mejoramiento en HbA1c no fue afectado por el género, edad, raza, etnicidad hispánica, BMI de línea base o dosis de pioglitazona de línea base. Un análisis pre-especificado por HbA1c de línea base demostró que los pacientes quienes entraron en el estudio con un HbA1c ≥8% lograron una reducción media significativa de la línea base de -1.1% sobre Incresina a 25 mg versus -0.3% con placebo a la Semana 26. En comparación al placebo, fueron también observadas reducciones clínicamente significativas en HbA1c con 25 mg de INCRESINA® no obstante de si los sujetos estaban recibiendo o no la terapia concomitante con metformina o sulfonilurea (-0.56% a -0.63%). No se observó diferencia significativa entre INCRESINA® y el placebo en el cambio de peso corporal cuando se administró en combinación con pioglitazona a la Semana 26. Los efectos lipídicos fueron también en general neutros.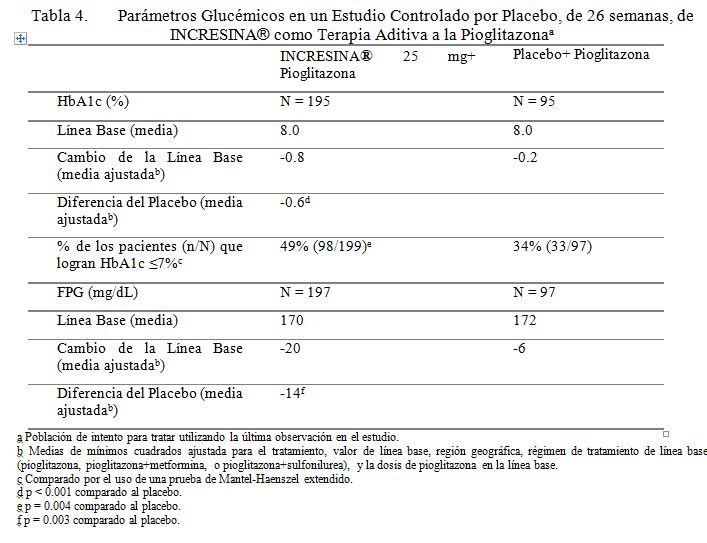
Terapia Aditiva a la Metformina: Se conduce un estudio controlado por placebo de 26 semanas para evaluar la eficacia y seguridad de INCRESINA® como terapia aditiva a la metformina en pacientes con diabetes tipo 2. Un total de 527 pacientes controlados inadecuadamente con metformina (HbA1c media de línea base = 8.0%) fueron repartidos aleatoriamente para recibir INCRESINA® 12.5 mg, INCRESINA® 25 mg, o placebo. Los pacientes fueron mantenidos en una dosis estable de metformina (dosis media = 1847 mg) durante el periodo de tratamiento. Todos los pacientes entraron a un periodo de corrida con placebo de ciego simple, de 4 semanas, antes de la repartición aleatoria. Después de la repartición aleatoria, todos los pacientes continuaron recibiendo instrucciones sobre la dieta y el ejercicio. Los pacientes quienes fallaron en cumplir las metas glucémicas definidas por el estudio, específicas, fueron rescatados y se les permitió entrar a un estudio de extensión con rótulos abiertos con INCRESINA®. La adición de 25 mg de INCRESINA® una vez al día a la terapia con metformina dio como resultado mejoramientos significativos de la línea base en HbA1c y FPG a la Semana 26, cuando se comparó a la adición del placebo (Tabla 5). También, significativamente nuevos pacientes que recibieron 25 mg de INCRESINA® (8%) requirieron rescate hiperglucémico en comparación a aquellos que recibieron placebo (24%) durante el estudio. El mejoramiento en HbA1c no fue afectado por el género, edad, raza, etnicidad hispánica, BMI de línea base o dosis de metformina de línea base. Un análisis pre-especificado por HbA1c de línea base, demostró que los pacientes quienes entraron en el estudio con un HbA1c ≥8% lograron una reducción media significativa de la línea base de -0.8% sobre INCRESINA ® a 25 mg versus -0.3% con placebo a la Semana 26. No se observó diferencia significativa entre Incresina y el placebo en el cambio de peso corporal cuando se administró en combinación con metformina a la Semana 26. Los efectos lipídicos fueron también en general neutros.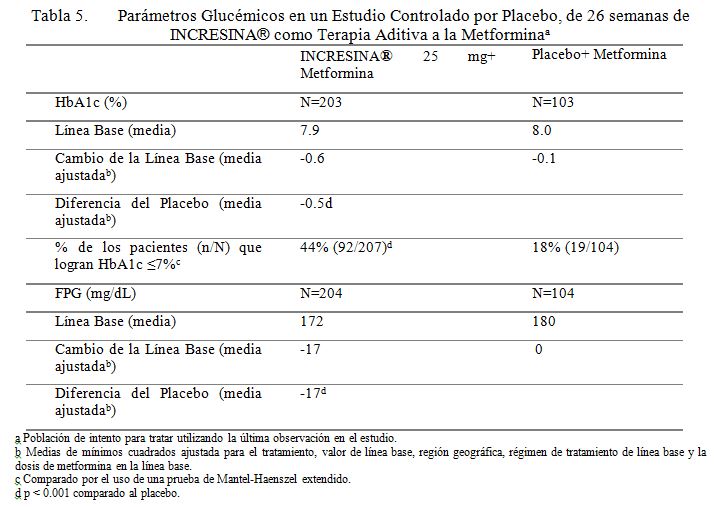
Terapia Aditiva a la Sulfonilurea: Se conduce un estudio controlado por placebo de 26 semanas para evaluar la eficacia y seguridad de INCRESINA® como terapia aditiva a la gliburida en pacientes con diabetes tipo 2. Un total de 500 pacientes controlados inadecuadamente con una sulfonilurea (HbA1c media de línea base = 8.1%) fueron repartidos aleatoriamente para recibir INCRESINA® 12.5 mg, Incresina 25 mg, o placebo. Los pacientes fueron mantenidos en una dosis estable de gliburida (dosis media = 12 mg) durante el periodo de tratamiento. Todos los pacientes entraron a un periodo de corrida con placebo de ciego simple, de 4 semanas, antes de la repartición aleatoria. Después de la repartición aleatoria, todos los pacientes continuaron recibiendo instrucciones sobre la dieta y el ejercicio. Los pacientes quienes fallaron en cumplir las metas glucémicas definidas por el estudio, específicas, fueron rescatados y se les permitió entrar a un estudio de extensión con rótulos abiertos con INCRESINA®. La adición de INCRESINA® a 25 mg una vez al día a la terapia con gliburida dio como resultado mejoramientos significativos de la línea base en HbA1c a la Semana 26, cuando se comparó a la adición del placebo (Tabla 6). Un cambio medio de la línea base en FPG a la Semana 26 para INCRESINA® a 25 mg mostró una reducción de 8 mg/dL en comparación a un incremento de 2 mg/dL con el placebo. Durante el estudio, significativamente menos pacientes que recibieron INCRESINA® a 25 mg (16%) requirieron rescate hiperglucémico en comparación a aquellos quienes recibieron placebo (28%). El mejoramiento en HbA1c no fue afectado por el género, edad, raza, etnicidad hispánica, BMI de línea base o dosis de gliburida de línea base. Un análisis pre-especificado por HbA1c de línea base, demostró que los pacientes quienes entraron en el estudio con un HbA1c ≥8% lograron una reducción media significativa de la línea base de -0.7% sobre INCRESINA® a 25 mg versus -0.1% con placebo a la Semana 26. No se observó diferencia significativa entre INCRESINA® y el placebo en el cambio de peso corporal cuando se administró en combinación con gliburida a la Semana 26. Los efectos lipídicos fueron también en general neutros.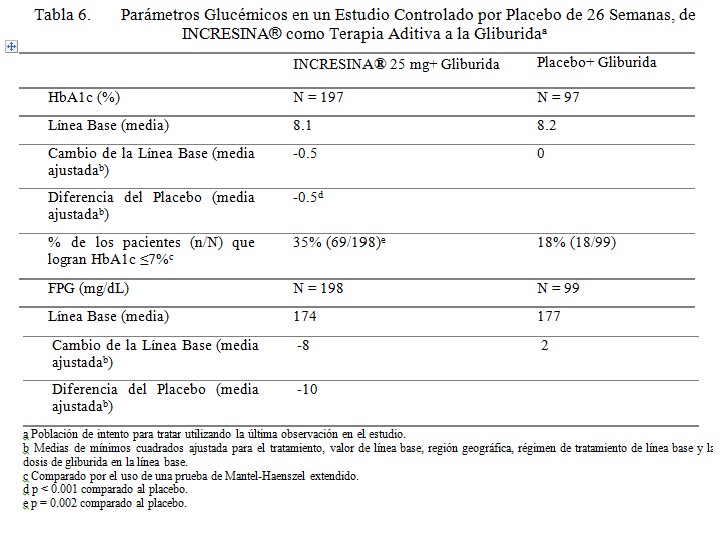
Terapia Aditiva a la Insulina: Se condujo un estudio controlado por placebo, de 26 semanas, para evaluar la eficacia y la seguridad de INCRESINA® como terapia aditiva a la insulina en pacientes con diabetes tipo 2. Un total de 390 pacientes controlados inadecuadamente con insulina sola o en combinación con metformina (media de línea base HbA1c = 9.3%) fueron repartidos aleatoriamente para recibir 12.5 mg de INCRESINA®, 25 mg de INCRESINA®, o placebo. Los pacientes fueron mantenidos en su régimen de insulina después de la repartición aleatoria y aquellos previamente tratados con insulina en combinación con metformina antes de la repartición aleatoria continuaron en el régimen de combinación durante el periodo de tratamiento. Todos los pacientes entraron a un periodo de corrida con placebo, de ciego simple, de 4 semanas, antes de la repartición aleatoria. Después de la repartición aleatoria, todos los pacientes continuaron recibiendo instrucciones sobre la dieta y el ejercicio. Los pacientes quienes fallaron en cumplir las metas glucémicas definidas por el estudio, específicas, fueron rescatados y se les permitió entrar a un estudio de extensión con rótulos a la vista, con INCRESINA®. La adición de 25 mg de INCRESINA® una vez al día a la terapia con insulina dio como resultado mejoramientos significativos de la línea base en HbA1c y FPG a la Semana 26, cuando se comparó a la adición del placebo (Tabla 7). También, significativamente nuevos pacientes que recibieron 25 mg de INCRESINA® (20%) requirieron rescate hiperglucémico en comparación a aquellos que recibieron placebo (40%) durante el estudio. El mejoramiento en HbA1c no fue afectado por el género, edad, raza, etnicidad hispánica, BMI de línea base o dosis de insulina de línea base. Un análisis pre-especificado por HbA1c de línea base, demostró que los pacientes quienes entraron en el estudio con un HbA1c ≥9% lograron reducciones significativas de la línea base de -0.8% sobre INCRESINA® a 25 mg versus -0.3% con placebo a la Semana 26. En comparación al placebo, fueron también observadas reducciones clínicamente significativas en HbA1c con 25 mg de INCRESINA®, no obstante de si los pacientes estaban o no recibiendo también terapia concomitante con metformina (-0.57% a -0.62%). No se observó diferencia significativa entre INCRESINA® y el placebo en el cambio de peso corporal cuando se administró en combinación con insulina a la Semana 26. Los efectos lipídicos fueron también en general neutros.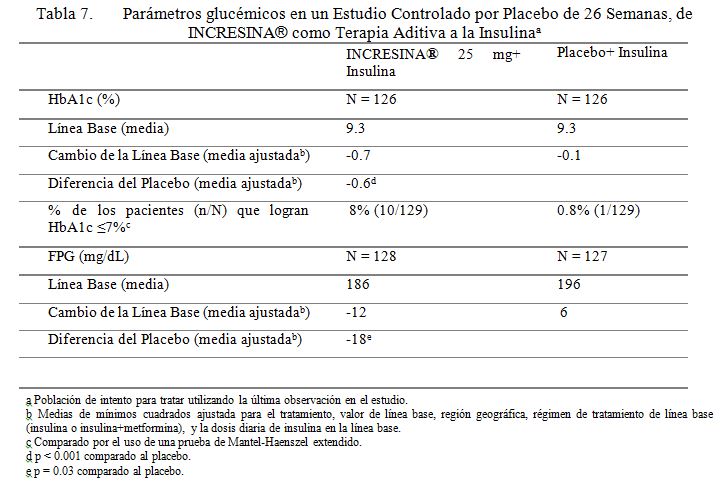
Estudios de comparador activo a largo plazo: Estudio multinacional, a doble ciego, aleatorio para evaluar la combinación de alogliptina más metformina comparado con glipizida más metformina durante 104 semanas. Se llevó a cabo un estudio multinacional (30 países, 310 sitios), a doble ciego, controlado con medicamento activo en pacientes con diabetes mellitus tipo 2 con control glucémico insuficiente, a pesar de un tratamiento con hidrocloruro de metformina (≥1500 mg). Un total de 2639 pacientes fueron distribuidos aleatoriamente a uno de tres grupos de tratamiento: alogliptina 12.5 mg, alogliptina 25 mg una vez al día o glipizida 5 mg una vez al día, titulada en hasta 20 mg una vez al día. Los pacientes se mantuvieron en una dosis estable de metformina durante el periodo de tratamiento. Los resultados del análisis mostraron que el tratamiento de metformina más alogliptina 12.5 mg y 25 mg no era inferior comparado con glipizida más metformina en concentraciones de HbA1c cada vez menores en las Semanas 52 y 104. La alogliptina 25 mg más metformina demostraron superioridad en comparación con glipizida más metformina en la Semana 104. Tanto en la Semana 52 como en la 104, la alogliptina dio mejores resultados en las medidas glucémicas secundarias (FPG, medidas de respuesta clínica, PPG a 2h e incidencia de rescate glucémico) en comparación con la glipizida; la alogliptina redujo significativamente el peso corporal en comparación con la glipizida. La alogliptina se asoció con una tasa de hipoglucemia mucho menor (10 veces menos) comparada con la glipizida en la Semana 104. La resultados finales (Semana 104) del estudio se presentan en la Tabla 8.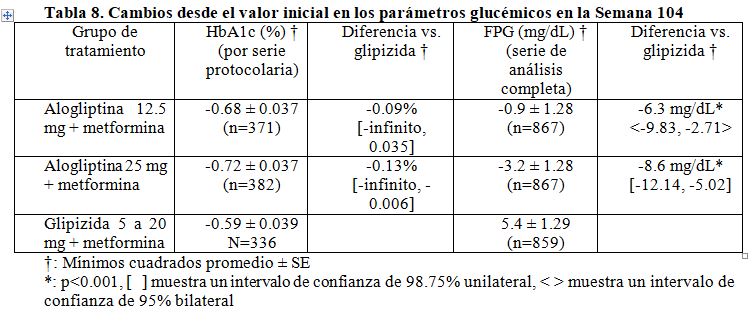
Seguridad cardiovascular: Se realizó un estudio prospectivo y aleatorio de seguridad CV con 5380 pacientes para analizar el efecto de la alogliptina en comparación con placebo (como complemento de los cuidados estándar) en eventos adversos cardiovasculares mayores (MACE), incluyendo el tiempo a la primera incidencia de cualquier evento relacionado con muerte CV, infarto al miocardio (IM) no fatal y derrame no fatal en pacientes con un evento coronario agudo reciente (15 a 90 días). En el valor inicial, los pacientes tenían una edad promedio de 61 años, una duración promedio de diabetes de 9.2 años y una HbA1c promedio de 8.0%. Los pacientes de este estudio corrían un mayor riesgo de presentar un evento CV con un historial de IM (88%), insuficiencia cardiaca congestiva (27.9%), angina inestable (31.1%), accidente cerebrovascular (CVA) (7.2%), hipertensión (83.1%), dislipidemias (57.2%) daño renal (moderado: 26.2%; severo/ESRD: 2.9%). El estudio mostró que la alogliptina no aumentó el riesgo de tener un MACE comparado con el placebo [Riesgo relativo (RR): 0.96; intervalo de confianza (IC) de 99% unilateral: 0-1.16]. En el grupo de alogliptina, 11.3% de los pacientes tuvieron un MACE en comparación con 11.8% de los pacientes en el grupo de placebo (Tabla 9).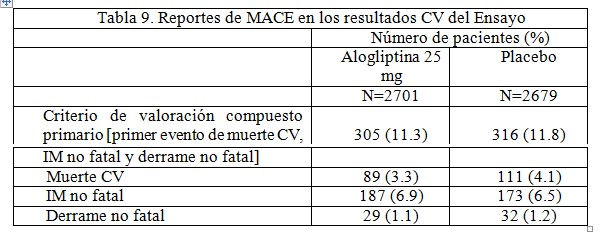
Hubo 703 pacientes que presentaron un evento dentro del criterio de valoración compuesto secundario en MACE (primer evento de muerte CV, IM no fatal, derrame no fatal y revascularización urgente debido a angina inestable). En el grupo de alogliptina, 12.7% (344 sujetos) presentaron un evento dentro del criterio de valoración secundario en MACE, en comparación con 13.4% (359 sujetos) en el grupo de placebo [RR=0.95; IC de 99% unilateral: 0-1.14]. En general durante el estudio, hubo 153 sujetos (5.7%) en el grupo de alogliptina y 173 sujetos (6.5%) en el grupo de placebo que murieron. De ellos, 112 pacientes (4.1%) en el grupo de alogliptina tuvieron muerte cardiovascular (incluyendo las que ocurrieron después de un primer evento de IM y/o derrame) comparado con 130 sujetos (4.9%) que recibieron placebo [RR = 0.851; IC de 95% bilateral: 0.662, 1.096].
Dosis y via de administración: Dosis Recomendada: La dosis recomendada de INCRESINA® es de 25 mg una vez al día, como monoterapia o como terapia en combinación. INCRESINA® puede ser tomado con o sin alimento. Pacientes con Insuficiencia Renal. Para pacientes con insuficiencia renal leve (depuración de creatinina [CrCl] ≥50 ml/min, que corresponde aproximadamente a los niveles de creatinina en suero de ≤1.7 mg/dL en hombres y ≤1.5 mg/dL en mujeres), no es necesario ningún ajuste de la dosis de INCRESINA®. Para pacientes con insuficiencia renal moderada (CrCl ≥30 a < 50 ml/min, que corresponde aproximadamente a los niveles de creatinina en suero de > 1.7 a ≤3.0 mg/dL en hombres y > 1.5 a ≤2.5 mg/dL en mujeres), la mitad de la dosis terapéutica de INCRESINA® debe ser administrada (12.5 mg una vez al día). Para pacientes con insuficiencia renal severa (CrCl < 30 ml/min, que corresponde aproximadamente a niveles de creatinina en suero de > 3.0 mg/dL en hombres y > 2.5 mg/dL en mujeres) o con Enfermedad Renal Terminal que requiere diálisis, un cuarto de la dosis terapéutica de INCRESINA ® debe ser administrada (6.25 mg una vez al día). INCRESINA® puede ser administrado sin considerar el tiempo de la diálisis. Debido a que existe una necesidad para el ajuste de dosis con base en la función renal, se recomienda la evaluación de la función renal antes del inicio de terapia con INCRESINA® y periódicamente después de éste. La depuración de la creatinina puede ser estimada a partir de la creatinina en suero utilizando la fórmula de Cockcroft-Gault. Insuficiencia hepática: No se requieren ajustes de dosis en pacientes con insuficiencia hepática leve o moderada (Child-Pugh Grado A y B o calificación 5 a 9). La alogliptina no ha sido estudiada en pacientes con insuficiencia hepática severa (Child-Pugh Grado C o calificación > 9), y por lo tanto, no se recomienda en estos pacientes. Uso Pediátrico: No ha sido establecida la seguridad de la efectividad de INCRESINA® en pacientes pediátricos. Uso Geriátrico: De un número total de pacientes (N = 2454) en los estudios de seguridad clínica y eficacia, tratados con INCRESINA®, 468 pacientes fueron 65 años y más, y 61 pacientes fueron 75 años de edad y más. No se observaron diferencias generales en la seguridad o en la efectividad entres esos pacientes y pacientes más jóvenes. Mientras que ésta y otras experiencias clínicas reportadas no han identificado diferencias en la respuesta entre los pacientes más viejos y más jóvenes, no puede ser excluida mayor sensibilidad de algunos individuos más viejos.
Manifestaciones y manejo de la sobredosificacion o ingesta accidental: Las dosis más altas de alogliptina que se administraron en los ensayos clínicos fueron dosis únicas de 800 mg en sujetos sanos y dosis múltiples de 400 mg una vez al día durante 14 días en sujetos con diabetes tipo 2 (equivalente a 32 veces y 16 veces la máxima dosis clínica recomendada de 25 mg). No se observaron eventos adversos serios en estos niveles de dosis. En caso de sobredosis, debe iniciarse el retiro de material no absorbido del tracto gastrointestinal e instituir el monitoreo clínico necesario y la terapia de apoyo según lo requiere el estado clínico del paciente. La alogliptina puede dializarse mesuradamente. Luego de 3 horas de hemodiálisis se retiró aproximadamente 7% del fármaco. Por lo tanto, es poco probable que la hemodiálisis sea útil en caso de sobredosis. Se desconoce si la alogliptina puede dializarse por medio de diálisis peritoneal.
Presentación(es): Frasco con 7, 14 ó 28 tabletas de 6.25 mg, 12.5 mg y 25 mg- Caja de cartón en envase de burbuja de 7, 14 ó 28 tabletas con 6.25 mg, 12.5 mg y 25 mg
Recomendaciones sobre almacenamiento: Consérvese a no más de 30°C, en lugar seco. Mantener la caja y el frasco bien cerrado. Manténgase en el empaque original.
Leyendas de protección: Literatura exclusiva para médico. Su venta requiere receta médica. Mantenga fuera del alcance de los niños. No se use durante el embarazo, la lactancia, ni en menores de 18 años. Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx.
Nombre y domicilio del laboratorio: Takeda México, S.A. de C.V. Av. Primero de Mayo No. 130. Col. Industrial Atoto C.P. 53519. Naucalpan de Juárez, México
Número de registro del medicamento: 163M2012 SSA IV