INCRUSE
GSK
Denominación genérica: Bromuro de Umeclidinio.
Forma farmacéutica y formulación: Polvo. Cada dosis contiene: Umeclidinio. Equivalentes a 65 mcg de bromuro de Umeclidinio 62.5 mcg. Excipiente cbp 12.5 mg.
Indicaciones terapéuticas: INCRUSE® está indicado en el tratamiento broncodilatador de manutención para aliviar síntomas asociados con enfermedad pulmonar obstructiva crónica (EPOC). INCRUSE® está indicado en pacientes con enfermedad pulmonar obstructiva crónica (EPOC) que persisten sintomáticos y/o con exacerbación en combinación con corticoesteroides inhalados / agonistas de los receptores beta2-adrenérgicos de acción prolongada (ICS/LABAs por sus siglas en inglés). INCRUSE® ha sido primariamente estudiado en combinación con furoato de fluticasona/vilanterol o propionato de fluticasona/salmeterol.
Farmacocinética y farmacodinamia: Farmacocinética: Absorción: Tras la administración por inhalación de umeclidinio en voluntarios sanos, la Cmax ocurrió a los 5 a 15 minutos. La biodisponibilidad absoluta de umeclidinio inhalado fue, en promedio, 13% de la dosis, con una contribución insignificante de la absorción oral. Después de la administración repetida de umeclidinio inhalado, se alcanzó el estado estable en los siguientes 7 a 10 días, con una acumulación de 1.5 a 2 veces mayor. La exposición sistémica a umeclidinio tras su administración inhalada fue proporcional a la dosis. Distribución: Después de la administración intravenosa a sujetos sanos, el volumen promedio de distribución fue de 86 litros. La unión a proteínas plasmáticas in vitro en plasma humano fue, en promedio, de 89%. Metabolismo: Estudios in vitro demostraron que umeclidinio experimenta un metabolismo mediado principalmente por la isoenzima CYP2D6 del citocromo P450, y es un substrato para el transportador de glicoproteína P (Pgp). Las principales vías metabólicas para umeclidinio son oxidativas (hidroxilación, O-desalquilación), seguidas de conjugación (glucuronidación, etc.), dando como resultado una variedad de metabolitos, ya sea con una actividad farmacológica disminuida, o para los que aún no se ha establecido la actividad farmacológica. La exposición sistémica a los metabolitos es baja. Interacciones farmacológicas: Umeclidinio es un sustrato del transportador de glicoproteína P (Pgp) y de CYP2D6. Se evaluó el efecto que ejerce el inhibidor del transportador de P-gp, verapamilo (240 miligramos una vez al día), en la farmacocinética en estado estable de umeclidinio en voluntarios sanos. No se observó que verapamilo ejerciera algún efecto en la Cmax de umeclidinio. Se observó un aumento de aproximadamente 1.4 veces en el AUC de umeclidinio. Se evaluó el efecto que produce la falta del genotipo metabolizador lento de CYP2D6 en el perfil farmacocinético en estado estable de umeclidinio en voluntarios sanos (metabolizadores normales de CYP2D6 y metabolizadores lentos de CYP2D6). No se observaron diferencias clínicamente significativas en la exposición sistémica a umeclidinio (500 microgramos), después de la administración por inhalación de dosis diarias repetidas a sujetos normales y metabolizadores lentos de CYP2D6. Eliminación: Después de la administración intravenosa, la depuración plasmática fue de 151 litros/hora. Tras la administración intravenosa, aproximadamente 58% de la dosis radiomarcada administrada (ó 73% de la radioactividad recuperada) se excretó en las heces a las 192 horas después de la dosis. La eliminación urinaria representó 22% de la dosis radiomarcada administrada a las 168 horas (27% de la radiactividad recuperada). Después de la administración intravenosa, la excreción de material relacionado con el fármaco en heces indicó la existencia de secreción con la bilis. Tras la administración oral a sujetos sanos de sexo masculino, la radiactividad total se excretó principalmente en heces (92% de la dosis radiomarcada administrada ó 99% de la radiactividad recuperada) a las 168 horas después de la dosis. Menos de 1% de la dosis administrada por vía oral (1% de la radiactividad recuperada) se excretó en orina, lo cual sugiere la existencia de una absorción insignificante después de la administración oral. La vida media de eliminación plasmática de umeclidinio tras su administración por inhalación durante 10 días fue, en promedio, de 19 horas, con una excreción de 3% a 4% del fármaco en forma inalterada en la orina en estado estable. Poblaciones especiales de pacientes: Personas de edad avanzada: Un análisis farmacocinético poblacional demostró que el perfil farmacocinético de umeclidinio es similar entre pacientes con EPOC de 65 años de edad y mayores, y aquellos menores de 65 años de edad. Insuficiencia renal: Los sujetos con insuficiencia renal grave (depuración de creatinina < 30 mililitros/min) no mostraron evidencia de un aumento en la exposición sistémica a umeclidinio (Cmax y AUC), y no hubo evidencia de una alteración en la unión a proteínas plasmáticas entre sujetos con insuficiencia renal grave y voluntarios sanos. Insuficiencia hepática: Los sujetos con insuficiencia hepática moderada (Clase B de Child-Pugh) no mostraron evidencia de aumento en la exposición sistémica a umeclidinio (Cmax y AUC), y no hubo evidencia de una alteración en la unión a proteínas plasmáticas entre sujetos con insuficiencia hepática moderada y voluntarios sanos. Umeclidinio no ha sido evaluado en sujetos con insuficiencia hepática grave. Otras características de pacientes: Un análisis farmacocinético poblacional demostró que no se requiere ajustar la dosis de umeclidinio, con base en el efecto de la edad, raza, género, uso de corticoesteroides inhalados o peso. Un estudio realizado en metabolizadores lentos de CYP2D6 no mostró evidencia de un efecto clínicamente significativo de polimorfismo genético de CYP2D6 en la exposición sistémica a umeclidinio. Estudios clínicos: Se evaluó la eficacia de INCRUSE® administrado una vez al día en dos estudios clínicos controlados con placebo, en pacientes adultos con un diagnóstico clínico de EPOC; un estudio de 12 semanas de duración (AC4115408) y un estudio de 24 semanas de duración (DB2113373). Estudios controlados con placebo: En el estudio de 12 semanas de duración, INCRUSE® demostró mejorías estadísticamente significativas y clínicamente importantes en las mediciones de la función pulmonar (tal como se definen por el cambio respecto al VEF1 basal en la semana 12, que fue el criterio primario de valoración de eficacia, en comparación con placebo (véase la Tabla 1). Los efectos broncodilatadores observados con INCRUSE, en comparación con el placebo, fueron evidentes después del primer día de tratamiento, y se mantuvieron durante el período de tratamiento de 12 semanas.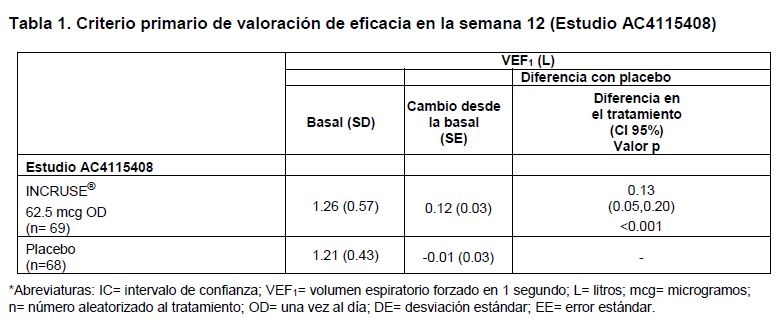
INCRUSE® demostró una mejoría estadísticamente significativa respecto al valor basal en el promedio ponderado del VEF1 durante 0-6 horas después de la administración en la semana 12, en comparación con el placebo (0.17 litros (p < 0.001)). El porcentaje de pacientes que recibieron INCRUSE® y que respondieron con una diferencia mínima clínicamente importante (MCID) de ≥1 unidad en el puntaje focal del Índice de Disnea Transicional (TDI) en la Semana 12, fue de 38% (24/64), en comparación con 15% (8/53) para el placebo. Las probabilidades de tener respuesta vs. no respuesta, de acuerdo con el TDI, fueron mayores de manera estadísticamente significativa para INCRUSE® en comparación con placebo, en la semana 12 (cociente de probabilidades de 3.4 (IC de 95% de 1.3, 8.4), p=0.009). INCRUSE® demostró mejorías estadísticamente significativas, en comparación con el placebo, en el cambio respecto al valor basal en el puntaje total en la semana 12 para el Cuestionario Respiratorio de St. George (SGRQ), una herramienta de medición específica del estado de salud (-7.90 unidades) (p < 0.001). El porcentaje de pacientes que recibieron INCRUSE® y que respondieron con una reducción ≥4 unidades (MCID) en el puntaje total del SGRQ en la semana 12, fue de 44% (28/63), en comparación con 26% (14/54) para el placebo. Las probabilidades de tener respuesta vs. no respuesta, de acuerdo con el SGRQ, fueron mayores de manera estadísticamente significativa para INCRUSE® en comparación con placebo, en la semana 12 (cociente de probabilidades de 2.44 (IC de 95% de 1.08, 5.50), p=0.032). Además, un número significativamente menor de pacientes tratados con INCRUSE® requirieron salbutamol de rescate durante el período de tratamiento de 12 semanas, en comparación con los tratados con placebo (reducción promedio de 0.7 atomizaciones por día, y la diferencia del placebo fue estadísticamente significativa (p=0.025)). En el estudio de 24 semanas de duración, INCRUSE® demostró mejorías estadísticamente significativas en la función pulmonar (tal como se definen por el cambio respecto al VEF1 basal en la semana 24, que fue el criterio primario de valoración de eficacia, en comparación con placebo (véase la Tabla 2). Los efectos broncodilatadores observados con INCRUSE®, en comparación con el placebo, fueron evidentes después del primer día de tratamiento, y se mantuvieron durante el período de tratamiento de 24 semanas.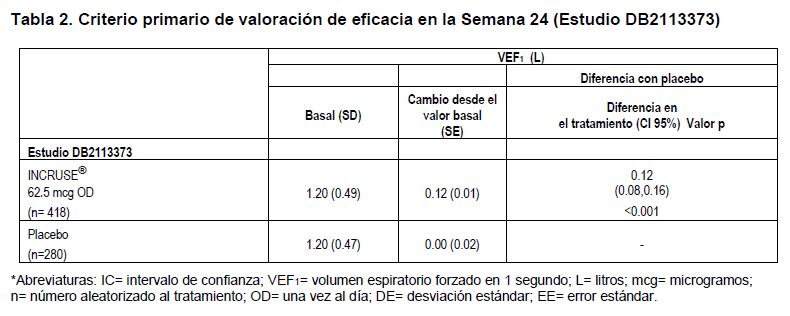
INCRUSE® demostró una mejoría estadísticamente significativa mayor respecto al valor basal en el promedio ponderado del VEF1 durante 0-6 horas después de la dosis en la semana 24, en comparación con el placebo (0.15 litros; (p < 0.001)). Se demostró una mejoría estadísticamente significativa respecto al placebo en el puntaje focal de TDI en la semana 24 para INCRUSE® (1.0 unidades) (p < 0.001). El porcentaje de pacientes que recibieron INCRUSE® y que respondieron con una diferencia mínima clínicamente importante (MCID) de ≥1 unidad en el puntaje focal de TDI en la semana 24, fue de 53% (207/394), en comparación con 41% (106/260) para placebo. Las probabilidades de tener respuesta vs. no tener respuesta, de acuerdo con el TDI, fueron mayores de manera estadísticamente significativa para INCRUSE® en comparación con placebo en la semana 24 (cociente de probabilidades de 1.6 (IC de 95% de 1.2, 2.3), p=0.002). Para INCRUSE® también se demostraron mejorías estadísticamente significativas, en comparación con el placebo, en el cambio respecto a la línea basal en el puntaje total en la semana 24 para el Cuestionario Respiratorio de St. George (SGRQ), una herramienta de medición específica del estado de salud (-4.69 unidades) (p£0.001). El porcentaje de pacientes que recibieron INCRUSE® y que respondieron con una reducción de ≥4 unidades (MCID) en el puntaje total del SGRQ en la semana 24, fue de 44% (172/388), en comparación con 34% (86/254) para el placebo. Las probabilidades de tener respuesta vs. no tener respuesta, de acuerdo con el SGRQ, fueron mayores de manera estadísticamente significativa para INCRUSE® en comparación con placebo, en la semana 24 (cociente de probabilidades de 1.6 (IC de 95% de 1.2, 2.3), p=0.003). El tratamiento con INCRUSE® disminuyó el riesgo de exacerbación de la EPOC, en comparación con el placebo (análisis del tiempo hasta la primera exacerbación; cociente de riesgo 0.6, IC 95%=0.4 a 1.0, reducción de riesgo de 40%). Estudios adicionales de eficacia conducidos con INCRUSE® en combinación con RELVARE® (furoato de fluticasona /vilanterol) 100/25 microgramos o SERETIDE® (propionato de fluticasona /salmeterol) 250/50 microgramos en pacientes adultos con diagnóstico clínico de EPOC: En dos estudios de 12 semanas, controlados con placebo (200109 y 200110), la adición de INCRUSE® a RELVARE® (100/25 microgramos) una vez al día, resultaron en mejoría estadísticamente significativa, e importante mejoría clínica en el objetivo primario VEF1, valle al día 85 comparado con placebo mas RELVARE® (124 mL (95% IC 93, 154, p < 0.001) y 122 mL (95% IC 91, 152, p < 0.001)). En dos estudios de 12 semanas, controlados con placebo (AC4116135 y AC4116136), la adición de INCRUSE® a SERETIDE® (250/50 microgramos) dos veces al día, resultaron en mejoría estadísticamente significativa, e importante mejoría clínica en el objetivo primario VEF1 valle al Día 85 comparado con placebo más SERETIDE® (147 mL (95% IC 107, 187, p < 0.001) y 127 mL (95%IC 89, 164, p < 0.001)). En estos estudios no se identificaron nuevas reacciones adversas debidas al medicamento con la adición de INCRUSE® a RELVARE® o SERETIDE®. Farmacodinamia: Mecanismo de acción: Umeclidinio es un antagonista de los receptores muscarínicos de acción prolongada (también denominado anticolinérgico). Es un derivado de quinuclidina, un antagonista de los receptores muscarínicos con actividad en múltiples subtipos de receptores colinérgicos muscarínicos. Umeclidinio ejerce su actividad broncodilatadora mediante la inhibición competitiva de la unión de acetilcolina con receptores colinérgicos muscarínicos en el músculo liso de las vías respiratorias. Presenta una baja reversibilidad in vitro en el subtipo M3 de receptores muscarínicos humanos, y acción de larga duración in vivo cuando se administra directamente en los pulmones en modelos pre-clínicos. Efectos farmacodinámicos: En un estudio de eficacia clínica controlado con placebo, de 24 semanas de duración, INCRUSE® aumentó el volumen espiratorio forzado en un segundo (VEF1) después de la administración de la primera dosis el día 1, con una mejoría de 0.07 litros a los 15 minutos, en comparación con el placebo (p < 0.001). El aumento desde el valor basal hasta el VEF1 máximo, durante las primeras 6 horas posteriores a la administración en el día 1, fue de 0.23 litros con INCRUSE®, en comparación con 0.11 litros para placebo. El aumento desde el valor basal hasta el VEF1 máximo, durante las primeras 6 horas posteriores a la administración en la semana 24, fue de 0.23 litros con INCRUSE® en comparación con 0.10 litros para placebo. Efectos cardiovasculares: En un estudio del QT, controlado con placebo y moxifloxacino, y realizado en 103 voluntarios sanos, se evaluó el efecto que ejerce umeclidinio 500 microgramos en el intervalo QT. Tras la administración de dosis repetidas de 500 microgramos de umeclidinio una vez al día durante 10 días, no se observaron efectos clínicamente relevantes en la prolongación del intervalo QT (corregido utilizando el método de Fridericia).
Contraindicaciones: INCRUSE® está contraindicado en pacientes con alergia a la proteína de la leche. Hipersensibilidad a los componentes de la fórmula.
Precauciones generales: INCRUSE® está indicado en el tratamiento de mantenimiento de la EPOC. No debe utilizarse para el alivio de síntomas agudos, es decir, como terapia de rescate para el tratamiento de episodios agudos de broncoespasmo. Los síntomas agudos deben ser tratados con un broncodilatador inhalado de acción corta. El uso cada vez más frecuente de broncodilatadores de acción corta para aliviar síntomas indica un deterioro en el control, por lo cual los pacientes deben ser examinados por un médico. Al igual que con otras terapias inhaladas, la administración de INCRUSE® puede producir broncoespasmo paradójico que podría poner en peligro la vida. En caso de ocurrir broncoespasmo paradójico, se debe suspender el tratamiento con INCRUSE® y, de ser necesario, instituir una terapia alternativa. Es posible que se observen efectos cardiovasculares tales como arritmias, p. ej., fibrilación auricular y taquicardia, después de la administración de antagonistas de receptores muscarínicos, incluyendo INCRUSE®. Por lo tanto, INCRUSE® debe utilizarse con precaución en pacientes con trastornos cardiovasculares graves, en especial con arritmias cardiacas. Consistentemente con su actividad antimuscarínica, INCRUSE® debe utilizarse con precaución en pacientes con glaucoma de ángulo cerrado o retención urinaria. Asma: El bromuro de umeclidinio no se debe utilizar en pacientes con asma, ya que no se ha estudiado en esta población de pacientes. Excipientes: Este medicamento contiene lactosa. Los pacientes con alteraciones hereditarias poco frecuentes de intolerancia a la galactosa, insuficiencia de lactasa de Lapp o malabsorción de glucosa o galactosa no deben utilizar este medicamento. Efectos en la capacidad de conducir y operar maquinaria: No se han realizado estudios para investigar el efecto que ejerce INCRUSE® en el desempeño automovilístico o la capacidad de operar maquinaria. No se han observado efectos adversos asociados con INCRUSE® ELLIPTA® que pudieran afectar la capacidad de desempeñar tareas que requieran discernimiento, o habilidades motrices o cognitivas.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: Existe una cantidad limitada de información sobre el uso de INCRUSE® en mujeres embarazadas. Los estudios realizados en animales no indican la existencia de efectos perjudiciales, ya sea directos o indirectos, con respecto a una toxicidad en la reproducción (véase Información no clínica). INCRUSE® sólo debe utilizarse durante el embarazo si el beneficio esperado para la madre justifica el posible riesgo para el feto. Lactancia: Se desconoce si umeclidinio se excreta en la leche materna. No es posible excluir un riesgo para neonatos/lactantes que se encuentran amamantando. Se debe tomar una decisión acerca de suspender la lactancia o suspender la terapia con INCRUSE®, tomando en consideración el beneficio de la lactancia para el niño y el de la terapia para la madre.
Reacciones secundarias y adversas:Información de estudios clínicos: Se evaluó el perfil de seguridad de umeclidinio a partir de aproximadamente 1,700 pacientes con EPOC que recibieron dosis de 62.5 microgramos o mayores hasta por un año. Esto incluye a aproximadamente 600 pacientes que recibieron la dosis recomendada de 62.5 microgramos una vez al día. En la siguiente tabla se presentan las reacciones adversas que se identificaron a partir de los cuatro estudios de eficacia y el estudio de seguridad a largo plazo (los cuales implicaron a aproximadamente 1,400 pacientes que recibieron umeclidinio). A continuación se presentan las reacciones adversas al medicamento (RAM) por clase de sistema orgánico del MedDRA y por frecuencia. Se ha utilizado la siguiente convención para la clasificación de las reacciones adversas: Muy comunes ≥1/10. Comunes ≥1/100 y < 1/10. Poco comunes ≥1/1,000 y < 1/100. Raras ≥1/10,000 y < 1/1,000. Muy raras < 1/10,000.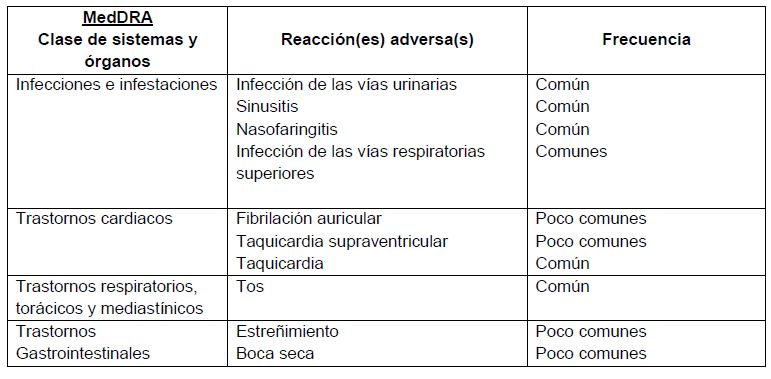

Interacciones medicamentosas y de otro género: La información clínica disponible no ha revelado la existencia de interacciones farmacológicas clínicamente pertinentes (véase Farmacología clínica). Otros agentes antimuscarínicos: No se ha estudiado la administración conjunta de bromuro de umeclidinio con otros antagonistas muscarínicos de acción prolongada u otros medicamentos que contengan este principio activo, por lo que no se recomienda su uso conjunto, ya que se podría potenciar los efectos adversos ya conocidos de los antagonistas muscarínicos inhalados.
Alteraciones en los resultados de pruebas de laboratorio: No hay información disponible.
Precauciones generales en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Datos de seguridad preclínica: En estudios no clínicos con umeclidinio, los hallazgos fueron aquellos típicamente relacionados con la farmacología primaria de antagonistas de receptores muscarínicos y/o irritación local. Fertilidad: No hay datos sobre los efectos de INCRUSE® en la fertilidad humana. Los estudios realizados en animales indican que INCRUSE® no ejerce efectos en la fertilidad (véase Información no clínica). Carcinogénesis/mutagénesis: Umeclidinio no fue genotóxico en un conjunto estándar de estudios, ni fue carcinogénico en estudios de inhalación de por vida en ratones o ratas a exposiciones ≥26 ó ≥22 veces mayores a la exposición clínica en seres humanos a 62.5 microgramos de umeclidinio, con base en el AUC, respectivamente. Toxicología en la reproducción: Umeclidinio no tuvo efectos adversos en la fertilidad masculina o femenina en ratas. Umeclidinio no fue teratogénico en ratas o conejos. En un estudio prenatal y postnatal, la administración subcutánea de umeclidinio a ratas dio lugar a menores aumentos de peso corporal materno y consumo de alimentos, y disminuyó ligeramente los pesos corporales de las crías previos al destete en hembras tratadas con una dosis de 180 microgramos/kg/día (aproximadamente 80 veces la exposición clínica en seres humanos a 62.5 microgramos de Umeclidinio, basado en el AUC).
Dosis y vía de administración: Vía de administración: Bucal. Consideración de uso: Para inhalación: INCRUSE® debe administrarse una vez al día, a la misma hora del día cada día. Cada dosis administrada (la dosis que sale de la boquilla del inhalador) contiene 55 microgramos de umeclidinio (equivalentes a 65 microgramos de bromuro de umeclidinio). Esto corresponde a una dosis pre-dispensada de 62.5 microgramos de umeclidinio (equivalentes a 74.2 microgramos de bromuro de umeclidinio). Adultos: La dosis recomendada consiste en una inhalación de INCRUSE® una vez al día. Niños: Debido a la indicación de este producto, no se recomienda en pacientes menores de 18 años de edad. Personas de edad avanzada: No se requiere ajustar la dosis en pacientes mayores de 65 años de edad (véase Farmacocinética - Poblaciones especiales de pacientes). Insuficiencia renal: No se requiere ajustar la dosis en pacientes con insuficiencia renal (véase Farmacocinética - Poblaciones especiales de pacientes). Insuficiencia hepática: No se requiere ajustar la dosis en pacientes con insuficiencia hepática leve o moderada. INCRUSE® no ha sido estudiado en pacientes con insuficiencia hepática grave (véase Farmacocinética - Poblaciones especiales de pacientes).
Manifestaciones y manejo de la sobredosificación o ingesta accidental: No se dispone de información obtenida de estudios clínicos en relación con la sobredosis de INCRUSE®. Síntomas y signos: Una sobredosis de INCRUSE® probablemente producirá signos y síntomas acordes a los efectos adversos conocidos de los antagonistas muscarínicos inhalados (p. ej., xerostomía, alteraciones en la acomodación visual y taquicardia). Tratamiento: En caso de sobredosis, el paciente debe recibir tratamiento complementario con monitoreo adecuado, según sea necesario. El manejo adicional deberá llevarse a cabo según lo indicado clínicamente, o de acuerdo con lo recomendado por el centro nacional de toxicología, donde esté disponible.
Presentaciones: Caja con dispositivo inhalador con 30 dosis.
Recomendaciones sobre almacenamiento: No almacenar a temperaturas superiores a 30°C. Si se almacena en un refrigerador, permita que el inhalador vuelva a la temperatura ambiente durante cuando menos una hora antes de su uso. Vida útil durante su uso: Después de retirarse la bandeja blíster, el producto puede almacenarse durante un período máximo de 6 semanas. Anote en el espacio proveído en la etiqueta la fecha en que debe desechar el inhalador. La fecha se debe agregar tan pronto como el inhalador se ha extraído de la bandeja.
Leyendas de protección: Su venta requiere receta médica No se deje al alcance de los niños Literatura exclusiva para médicos. Reporte las sospechas de reacción adversa a los correos: farmacovigilancia@cofepris.gob.mx y farmacovigilancia.mx@gsk.com. No se use durante el embarazo o lactancia. No se use en menores de 18 años.
Nombre y domicilio del laboratorio: GlaxoSmithKline México, S.A. de C.V. Calz. México Xochimilco No. 4900, Col. San Lorenzo Huipulco, C.P. 14370, Deleg. Tlalpan, Ciudad de México, México.
Número de registro del medicamento: 236M2015 SSA IV
Clave IPPA: GDS 09 / IPI 10 07 febrero 2017 / Actualización: 15 de mayo 2017.