INVIRASE®
ROCHE
Denominación genérica: Saquinavir.
Forma farmacéutica y formulación: Comprimidos. Cada comprimido contiene: mesilato de saquinavir equivalente a 500 mg de saquinavir. Excipiente cbp 1 comprimido.
Indicaciones terapéuticas: INVIRASE® está indicado para el tratamiento de pacientes adultos infectados por el VIH-1. INVIRASE® siempre debe administrarse en asociación con ritonavir y otros fármacos antirretrovirales.
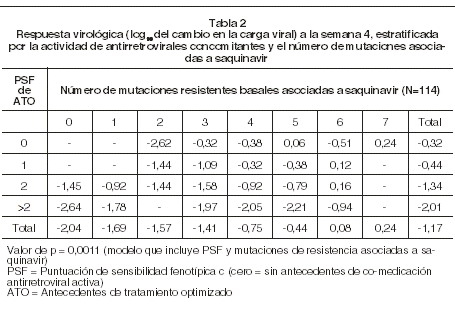
Farmacocinética y farmacodinamia: Farmacodinamia: mecanismo de acción: la proteasa del VIH es una enzima viral esencial necesaria para la división específica de las poliproteínas virales gag l y gag-pol. Estas poliproteínas virales contienen un tipo de sitio de separación el cual es reconocido sólo por el VIH y relacionado cercanamente con proteasas virales. Saquinavir ha sido diseñado como una estructura tipo péptido mimética del sitio de separación viral. El saquinavir es un inhibidor selectivo y reversible de la proteasa del VIH que impide la creación de partículas virales maduras e infecciosas. Actividad antiviral in vitro: saquinavir demuestra actividad antiviral contra cepas de laboratorio y aislamientos clínicos de VIH-1 con valores típicos de CE50 y CE90 en el rango de 1-10 nM y 5-50 nM, respectivamente, utilizando líneas de células T infectadas de forma aguda o primaria de los linfocitos/monocitos humanos. La actividad antiviral in vitro se ha observado frente a un panel de aislamientos de VIH-1 grupo M subtipo B no ramificados (A, AE, C, D, F, G y H) y VIH-2 con valores de CE50 que van de 0,3-2,5 nM. En la presencia de 50% de suero humano o alfa-1 glicoproteína ácida (1 mg/ml), la actividad antiviral de saquinavir disminuye por un factor promedio de 25 veces y 14-veces, respectivamente. Resistencia in vitro: la selección in vitro de la resistencia del VIH-1 de tipo salvaje: las mutaciones reportadas con más frecuencia, G48V y L90M, se observaron para desarrollo in vitro de VIH-1 tipo salvaje, en presencia de mayores concentraciones de saquinavir. El virus recombinante que alberga mutaciones G48V y L90M exhibieron 7,9 veces y 3,3 veces reducciones en la susceptibilidad viral a saquinavir, respectivamente. Otras mutaciones en la proteasa como M36I, I54V, K57R, L63V se desarrollaron con menor frecuencia en presencia de saquinavir. Resistencia in vivo: pacientes sin tratamiento previo (naïve): cuatro estudios han investigado saquinavir potenciado con ritonavir en pacientes naïve [(saquinavir/ritonavir 1.600 mg/100 mg de ritonavir una vez al día (n = 349); 1.000 mg/100 mg dos veces al día (n = 92)]. El análisis de resistencia basal se realizó en 26 pacientes que experimentaron rebote virológico. Los datos de dos pacientes fueron excluidos, ya sea porque estaban presentes mutaciones basales a IP o una mutación firme en la proteasa (D30N) asociado con otro IP desarrollado posteriormente. Los virus de dos pacientes (2/24) desarrollaron mutaciones en la proteasa (M36I y M46i/m, respectivamente). Estas mutaciones no son típicamente asociadas con resistencia a saquinavir. No se observaron mutaciones en la proteasa asociada a saquinavir, tras la falla virológica. Pacientes con tratamientos previos: se evaluó el genotipo basal y durante el tratamiento en 22 pacientes pretratados con IP, quienes experimentaron falla virológica después de recibir régimen de saquinavir reforzado con ritonavir (estudios MaxCmin 1 y 2; 1.000/100 mg dos veces al día, n = 171). Los virus de ocho pacientes (8/22; 36%) desarrollaron mutaciones adicionales en la proteasa después de la falla virológica. La incidencia relativa de cada mutación fue: I84V (n = 4, 18%); F53L, A71V o G73S (n = 2, 9%); L10V, M46I, I54V, V82A o L90M (n = 1), 4,5%). Actividad antiviral según el genotipo y fenotipo basal:< los valores clínicos de corte genotípicos y fenotípicos que predicen la eficacia clínica de saquinavir potenciado con ritonavir se han obtenido de los análisis retrospectivos de los estudios RESIST 1 y 2 y análisis de una cohorte grande de hospital. El fenotipo basal de saquinavir (cambio en la susceptibilidad relativa para la referencia, Ensayo Pheno Sense) mostró ser un factor predictor de los resultados virológicos. Primero se observó que la respuesta virológica descendía cuando el rango del cambio excedía 2,3 veces, mientras que no se observó beneficio virológico cuando el rango de cambio excedía 12 veces. Un estudio clínico de cohorte (Marcelin et al., 2007) identificó nueve codones de proteasa (L10F/I/M/R/V, I15A/V, K20I/M/R/T, L24I, I62V, G73S/T, V82A/F/S/T, I84V, L90M) que se asociaron con disminución de la respuesta virológica para saquinavir/ritonavir (1.000/100 mg dos veces al día) en 138 pacientes naïve a saquinavir. La presencia de 3 o más mutaciones se asoció con una respuesta reducida a saquinavir/ritonavir. Para confirmar la asociación entre el número de estas mutaciones de resistencia asociadas a saquinavir y respuesta virológica usando un juego de datos independientes, la asociación se investigó utilizando datos para pacientes que recibieron saquinavir reforzado con ritonavir en el estudio clínico RESIST 1 y 2. Los estudios RESIST 1 y 2 enrolaron una población de pacientes pretratados extensamente, que incluía un 54% de pacientes tratados previamente con saquinavir. Este análisis confirmó la asociación entre el número de mutaciones asociadas a saquinavir (p = 0,0133, ver Tabla 1). Además, la mutación G48V, previamente identificada in vitro como una mutación a saquinavir, se presentó a niveles basales en virus de tres pacientes, ninguno de los cuales respondieron a la terapia. La respuesta virológica a la terapia HAART se basa en la actividad de los componentes individuales antirretrovirales. La relación entre el número de mutaciones basales a saquinavir y la actividad concomitante de los componentes antirretrovirales del régimen se validaron usando datos del nivel basal de actividad fenotípica. La asociación entre el número de mutaciones basales a saquinavir asociadas a la resistencia y la respuesta fue significativamente alta cuando la actividad del esquema optimizado fue considerado cuenta (p = 0,0011, ver Tabla 2). Los pacientes que recibieron saquinavir en presencia de terapia concomitante activa antirretroviral y que tenían menos mutaciones asociadas a saquinavir tuvieron una mejoría en la respuesta comparada con los pacientes que recibían menor comedicación activa y números más altos de mutaciones asociadas a saquinavir.
Eficacia / estudios clínicos: se han evaluado los efectos de saquinavir asociado a la zalcitabina (con o sin zidovudina) sobre el desenlace clínico y los marcadores biológicos (recuento de linfocitos CD4 y ARN plasmático) en pacientes infectados por el VIH-1, con o sin tratamiento antirretroviral previo. En un amplio estudio clínico abierto y aleatorizado (MaxCmin 1), llevado a cabo en pacientes adultos infectados por el VIH-1, se estudiaron los efectos de la cápsula de gelatina blanda de saquinavir (1.000 mg) en combinación con ritonavir (100 mg) administrado dos veces al día. En un estudio (NV14256), efectuado con pacientes previamente tratados con zidovudina (CD4 ≥ 50 ≤ 300 células/mm3), el tiempo hasta la aparición de la primera enfermedad definitoria de SIDA o de muerte fue mayor en los pacientes tratados con saquinavir y zalcitabina que en quienes recibieron sólo zalcitabina. La terapia en combinación disminuyó en un 53% el riesgo de que un paciente falleciera o presentara una enfermedad definitoria de SIDA; en cuanto al riesgo de muerte, la biterapia consiguió reducirlo en un 72%. Estas cifras corresponden a una reducción de la tasa de enfermedades definitorias de SIDA o muerte del 29,4% a un 16,0% durante un período de 18 meses; en cuanto al riesgo de muerte durante 18 meses, la tasa disminuyó del 8,6% al 4,1%. En los tres grupos, la duración mediana del tratamiento fue de 11 a 13 meses, con una mediana de seguimiento de 17 meses. En este estudio, la mediana de la cifra basal de linfocitos CD4 para todos los grupos de tratamiento osciló entre 156 y 176 células/mm3. El cambio promedio con respecto a los valores basales a lo largo de 16 semanas (DAVG16) con saquinavir y zalcitabina fue de +26/mm3 para los linfocitos CD4 y de -0.6 log10 para el número de copias de ARN por ml de plasma. El mayor aumento de la cifra de linfocitos CD4 fue de 47 células/mm3, en la semana 16. La reducción máxima de la viremia fue de 0,7 log10 copias/ml, en la semana 12. En el estudio de fase III SV14604, de diseño aleatorizado, multicéntrico paralelo y doble-ciego, se compararon la terapia con zidovudina y zalcitabina contra la terapia con saquinavir + zidovudina, vs. terapia con saquinavir + zidovudina + zalcitabina en pacientes infectados por el VIH con escaso o nulo tratamiento antirretroviral previo. Se suspendió prematuramente un cuarto grupo terapéutico con zidovudina en monoterapia; los pacientes inicialmente incluidos en él fueron modificados para recibir triterapia con saquinavir+ zidovudina + zalcitabina, y constituyeron un grupo con terapia triple "tardía". En total, 3.485 pacientes recibieron tratamiento y disponían de datos de seguimiento (población de intención de tratamiento). La mediana basal de linfocitos CD4 en los tres grupos terapéuticos osciló entre 199 - 204/ mm3, y la mediana basal de copias de ARN del VIH/ ml de plasma fue de 5,0 - 5,1 log10. La mediana de duración del tratamiento fue de 14 meses, con una mediana de seguimiento para enfermedades definitorias de SIDA o muerte de aproximadamente 17 meses. La progresión hasta la primera enfermedad definitoria de SIDA o muerte fue significativamente menor entre los pacientes con saquinavir + zidovudina + zalcitabina (76 casos, frente a los 142 registrados en el grupo con zidovudina + zalcitabina; p = 0,0001). Una comparación exploratoria entre el grupo con terapia triple "inicial" y el grupo con terapia triple "tardía" puso de manifiesto la superioridad de la terapia triple inicial con saquinavir (76 casos de enfermedad definitoria de SIDA o muerte, frente a 116 en el grupo con zidovudina inicial y terapia triple tardía; p = 0,001). El aumento del número de linfocitos CD4 fue mayor entre los pacientes tratados con terapia triple (mediana del aumento máximo con respecto a los valores basales: 71 células/ mm3) que en los que recibieron zidovudina + zalcitabina (40 células/ mm3). De forma similar, la disminución de la viremia fue mayor en el grupo con terapia triple (mediana del descenso máximo con respecto a los valores basales de copias de ARN del VIH por ml: -1,5 log10) que en el tratado con zidovudina + zalcitabina (-1,1 log10). Tanto para el recuento de linfocitos CD4 como para la viremia, las comparaciones durante las 48 semanas de seguimiento entre el grupo con terapia triple y el grupo con zidovudina + zalcitabina alcanzaron significación estadística (p = 0,0001). Con INVIRASE® en monoterapia únicamente se ha demostrado actividad antiviral escasa y pasajera. Por lo tanto, INVIRASE® debe asociarse siempre a otros antirretrovirales. En el estudio MaxCmin 1, la seguridad y la eficacia de la cápsula de gelatina blanda de saquinavir /ritonavir en dosis de 1.000/100 mg dos veces al día administrados con 2 ITRAN/ITRNN se compararon con las de indinavir/ritonavir en dosis de 800/100 mg dos veces al día junto con 2 ITRAN/ITRNN. En el grupo tratado con saquinavir/ritonavir, la mediana basal del recuento de linfocitos CD4 fue de 272 células/ mm3, y la mediana basal de copias de ARN del VIH por ml de plasma, de 4,0 log10 en el grupo tratado con indinavir/ritonavir, la mediana basal del recuento de linfocitos CD4 fue de 280 por mm3, y la mediana basal de copias de ARN del VIH por ml de plasma, de 3,9 log10. A la semana 48, el aumento de la cifra de linfocitos CD4 fue de 85 y 73 por mm3 en los grupos de saquinavir e indinavir respectivamente. Al realizar el análisis por intención de tratamiento (cambio = fracaso) en la semana 48, la proporción de pacientes con una viremia por debajo del límite de detección ( < 400 copias/ml) fue del 69% (n = 102) en el grupo con saquinavir frente al 53 % en el grupo con indinavir. En el estudio MaxCmin2, la seguridad y eficacia la de cápsula de gelatina blanda de saquinavir /ritonavir en dosis de 1.000/100 mg dos veces al día administrados junto con 2 ITRAN/ITRNN se compararon con las de lopinavir/ritonavir en dosis de 400/100 mg dos veces al día junto con 2 ITRAN/ITRNN en 324 sujetos. En el grupo tratado con saquinavir/ritonavir, la mediana basal del recuento de linfocitos CD4 fue de 241 /mm3, y la mediana basal de copias de ARN del VIH por ml de plasma, de 4,4 log10. En el grupo tratado con lopinavir/ritonavir, la mediana basal del recuento de linfocitos CD4 fue de 239 por mm3 y la mediana basal de copias de ARN del VIH por ml de plasma, de 4,6 log10. A las 48 semanas, la proporción de sujetos con una viremia por debajo del límite de detección ( < 50 copias/ml) fue del 53% (n = 161) en el grupo con saquinavir frente al 60% (n = 163) en el grupo con lopinavir al realizar el análisis por intención de tratamiento (cambio = fracaso), mientras que en el análisis en tratamiento (p = no significativo en ambas comparaciones) la proporción de sujetos con una viremia por debajo del límite de detección fue del 74% (n = 114) en el grupo con saquinavir frente al 70% (n = 141) en el grupo con lopinavir. La combinación de saquinavir y ritonavir puso de manifiesto una actividad virológica comparable a la del grupo con lopinavir y ritonavir cuando el cambio del tratamiento asignado se contabilizó como fracaso terapéutico. En el transcurso de 48 semanas se observó una fuerte respuesta inmunológica similar en ambos grupos, con un aumento mediano del recuento de linfocitos CD4 de 106 por mm3 en el grupo con lopinavir/ritonavir y de 110 por mm3 en el grupo con saquinavir/ritonavir. No se observaron diferencias en la incidencia de acontecimientos adversos de Grados 3 y 4 entre ambos grupos. Efectos sobre electrocardiograma: el efecto de 1.000/100 mg dos veces al día (dosis terapéutica) y 1.500/100 mg dos veces al día (dosis supra-terapéuticas) de INVIRASE®/ritonavir en el intervalo QT se evaluó durante 20 horas al día 3 de la dosis en un estudio cruzado de 4 vías, doble ciego, controlado con placebo y con control activo (moxifloxacino 400 mg), en voluntarios sanos hombres y mujeres, de 18 a 55 años (N = 59). La evaluación puntual en el día 3 fue elegida debido a que la exposición farmacocinética fue máxima en ese día en un estudio previo farmacocinético de dosis múltiple de 14 días. Estas dosis de INVIRASE®/ritonavir en el día 3 en este estudio dio lugar a una Cmáx media de aproximadamente 3 veces y 4 veces, respectivamente, superiores a la media de Cmáx observada con INVIRASE®/ritonavir 1.000/100 mg dos veces al día en la población de pacientes con VIH en estado estacionario. En el día 3, el intervalo de confianza del 95% superior de 1 cola de la diferencia media máxima en el QTcS corregido predosis al inicio (QT corregido de la frecuencia cardíaca específico del estudio) entre los brazos del fármaco activo y placebo fue > 10 mseg para los dos grupos de INVIRASE® reforzado con ritonavir (ver Tabla 3). La dosis supraterapéuticas de INVIRASE®/ritonavir parecieron tener un mayor efecto sobre el intervalo QT que la dosis terapéutica de INVIRASE®/ritonavir. La mayoría (89% y el 80% de la dosis terapéutica y la dosis supra-terapéuticas, respectivamente) de los sujetos tenían la QTcS de < 450 mseg y ninguno tenía el intervalo QTc de > 500 mseg.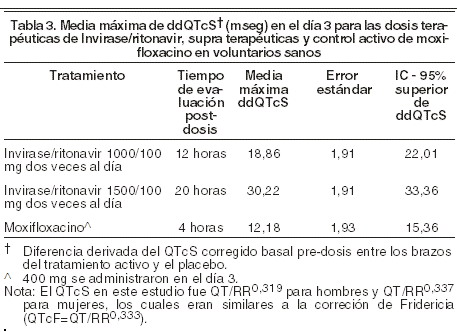
En este estudio, la prolongación de intervalo PR > 200 mseg también se observó en 40% y 47% de los sujetos que recibieron INVIRASE®/ritonavir 1.000/100 mg dos veces al día y en el grupo de 1.500/100 mg dos veces al día, respectivamente, en el día 3. Tres (3) % de los sujetos en el grupo de control activo con moxifloxacino y el 5% en el grupo placebo experimentaron prolongación del PR > 200 mseg. La media máxima de los cambios del intervalo PR en relación con el valor basal predosis fueron de 25 mseg y 34 mseg en los dos grupos de tratamiento de INVIRASE® reforzado con ritonavir, 1.000/100 mg dos veces al día y en el de 1.500/100 mg dos veces al día, respectivamente. No se observaron torsade de pointes ni prolongación del QT > 500 mseg en el estudio. En algunos sujetos, no se pudo descartar la asociación entre síncope o pre-síncope con prolongación de PR. La significancia clínica de estos hallazgos a partir de este estudio en voluntarios sanos con el uso de INVIRASE®/ritonavir en pacientes infectados por HIV no es clara, sin embargo, se deben evitar dosis de INVIRASE®/ritonavir que excedan 1.000/100 mg dos veces al día. Farmacocinética: absorción: en voluntarios sanos, el grado de absorción (determinado a partir del ABC) tras una dosis oral de 600 mg de saquinavir aumentó de 24 ng•h/ml (CV = 33%) en ayunas a 161 ng•h/ml (CV = 35%) cuando el saquinavir se administraba tras un desayuno copioso (48 g de proteínas, 60 g de hidratos de carbono, 57 g de grasas; 1.006 kcal). En presencia de alimentos aumentaron también el tmáx (tiempo transcurrido hasta la concentración plasmática máxima), de 2,4 a 3,8 horas, y, sobre todo, la Cmáx (concentración plasmática máxima), de 3,0 a 35,5 ng/ml. Se ha demostrado que el efecto de los alimentos persiste hasta 2 horas. Por lo tanto, INVIRASE® debe administrarse en las dos horas siguientes a una comida. En un estudio cruzado de 22 pacientes infectados con VIH tratados con INVIRASE®/ritonavir 1.000 mg/100 mg dos veces por día y recibiendo tres dosis consecutivas en condiciones de ayuno, o después de una comida alta en grasa y en calorías (46 g de grasa, 1.091 kcal), el ABC0-12 de saquinavir fue de 10320 ng•h/ml y de 34926 ng•h/ml, respectivamente. Todos los pacientes, excepto uno, alcanzaron el Cvalle por encima del umbral terapéutico en ayuno. Sin embargo, la combinación INVIRASE®/ritonavir debe administrarse dentro de las dos horas siguientes a una comida. La biodisponibilidad absoluta en promedio fue del 4% (rango: 1 - 9%) en 8 voluntarios sanos que recibieron una sola dosis de 600 mg de saquinavir tras un desayuno copioso. Esta escasa biodisponibilidad se atribuye a la asociación de una absorción incompleta y un notable metabolismo de primer paso hepático. Se ha demostrado que el pH gástrico apenas influye en el notable aumento de la biodisponibilidad cuando INVIRASE® se administra con los alimentos. Tras dosis orales múltiples (25-600 mg tres veces al día) en presencia de alimentos, el aumento de la exposición al fármaco (50 veces mayor) fue más que proporcional con respecto al aumento de la dosis (24 veces mayor). Tras la administración de dosis múltiples (600 mg tres veces al día) a pacientes seropositivos, el ABC en equilibrio fue 2,5 veces mayor (IC del 95%: 1,6 - 3,8) que la observada tras una dosis única. En los pacientes seropositivos a los que se administraron tres dosis diarias de 600 mg de saquinavir con instrucciones de tomarlas después de las comidas, el ABC y la Cmáx de saquinavir eran el doble de las obtenidas en los voluntarios sanos sometidos a idéntica dosis terapéutica (ver Tabla 4).
En los pacientes infectados por VIH, la cápsula de gelatina blanda de saquinavir o INVIRASE® en combinación con ritonavir en dosis de 400/400 mg dos veces al día o 1.000/100 mg dos veces al día proporcionan durante períodos superiores a 24 horas exposiciones sistémicas al saquinavir similares o mayores que las obtenidas con dosis de 1.200 mg de la cápsula de gelatina blanda de saquinavir tres veces al día (ver Tabla 5).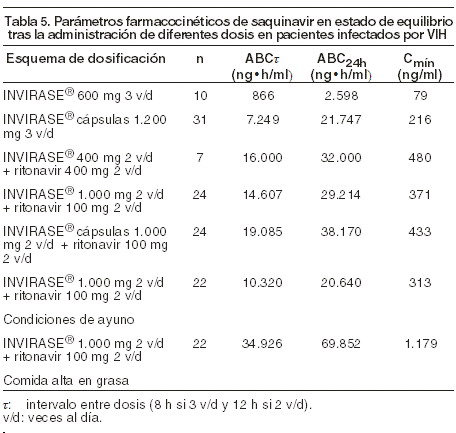
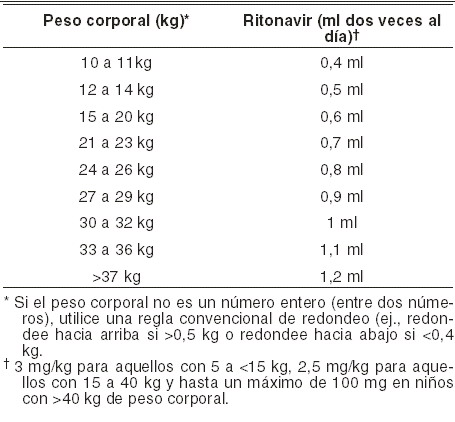
No se han observado diferencias en cuanto a absorción gastrointestinal entre los seropositivos con diarrea y sin ella; tampoco la administración de saquinavir afectó a esta variable. Saquinavir actúa de sustrato del transportador MDR1 transportador multidroga 1 (glucoproteína P, P-gp). Se ha demostrado la bioequivalencia de INVIRASE® Comprimidos (500 mg) e INVIRASE® Cápsulas de 200 mg en 94 voluntarios sanos de ambos sexos que recibieron 1.000 mg (2 x 500 mg) de INVIRASE® o (5 x 200 mg) de INVIRASE® con alimentos en combinación con 100 mg de ritonavir dos veces al día. Se estimó que el cociente de exposición media del saquinavir era de 1,10 para el ABC0- y de 1,19 para la Cmáx de saquinavir, con un intervalo de confianza del 90% de 1,04 - 1,16 y 1,14 - 1,25, respectivamente. Distribución: saquinavir se distribuye ampliamente en los tejidos. Tras la administración intravenosa de una dosis de 12 mg de saquinavir, el volumen de distribución en equilibrio fue de 700 litros (CV: 39%). El grado de unión del saquinavir a las proteínas plasmáticas es alto (98%, aproximadamente) e independiente de la concentración desde los 15 hasta los 700 ng/ml. En dos pacientes tratados con tres dosis diarias de 600 mg de INVIRASE®, las concentraciones de saquinavir en el líquido cefalorraquídeo fueron insignificantes en comparación con las correspondientes concentraciones plasmáticas. Metabolismo: estudios in vitro con microsomas de hígado humano han mostrado que el metabolismo de saquinavir está mediado por el sistema del citocromo P-450, siendo la isoenzima específica CYP3A4 responsable de más del 90% del metabolismo hepático. Según los estudios in vitro, saquinavir se metaboliza rápidamente a diversos compuestos inactivos monohidroxilados y dihidroxilados. Tras la administración intravenosa, el 66% del saquinavir circulante se encontraba en forma inalterada, y el resto, en forma de metabolitos. Estos resultados parecen indicar que el saquinavir sufre un considerable metabolismo de primer paso hepático. La depuración sistémica de saquinavir es elevada (1,14 l/h/kg; CV: 12%), ligeramente superior al flujo plasmático hepático; esta depuración es constante tras la administración intravenosa de 6, 36 y 72 mg de saquinavir. El tiempo medio de permanencia de saquinavir fue de 7 horas. Eliminación: en un estudio de balance de masas con 600 mg de saquinavir marcado con 14C (n = 8), el 88% de la radiactividad administrada por vía oral se recuperó en las heces durante los 4 primeros días, y sólo el 1% en la orina. En otros cuatro sujetos, tras la administración intravenosa de 10,5 mg de saquinavir radiomarcado con 14C, el 81% de la radiactividad administrada se eliminó por las heces durante los primeros 4 días, y el 3% por la orina. En los estudios de balance de masas tras administración oral, el 13% del saquinavir plasmático circulante se encontraba en forma inalterada, y el resto, en forma de metabolitos. Farmacocinética en poblaciones especiales: pacientes con insuficiencia renal: no se han realizado estudios farmacocinéticos de INVIRASE® en pacientes con insuficiencia renal. Pacientes con insuficiencia hepática: el efecto de la insuficiencia hepática sobre la farmacocinética en estado estacionario de saquinavir/ ritonavir (1.000 mg/100 mg dos veces durante 14 días) fue investigado en 7 pacientes infectados por VIH con insuficiencia hepática moderada (Child Pugh Grado B puntuación de 7 a 9). El estudio incluyó un grupo control de 7 pacientes infectados por VIH con función hepática normal empatados con los pacientes con insuficiencia hepática por edad, sexo, peso y consumo de tabaco. La media (% coeficiente de variación entre paréntesis) los valores de saquinavir ABC0-12 y Cmáx fueron 24,3 (102%) mg•h/ml y 3,6 (83%) mg•h/ml, respectivamente, para los pacientes infectados por VIH con insuficiencia hepática moderada. Los valores correspondientes en el grupo control fueron 28,5 (71%) mg•h/ml y 4,3 (68%) mg•h/ml. La relación media geométrica (cociente de los parámetros farmacocinéticos en pacientes con insuficiencia hepática a los pacientes con función hepática normal) (intervalo de confianza 90%) fue 0,7 (0,3 a 1,6) para ambos ABC0-12 y la Cmáx, lo que sugiere aproximadamente un 30% de reducción en la exposición farmacocinética en pacientes con insuficiencia hepática moderada. No hay justificación para el ajuste de la dosis de saquinavir en pacientes infectados por VIH con insuficiencia hepática moderada. Efectos del género, raza y edad: género: no se observó ningún efecto en función del género sobre la farmacocinética de las cápsulas de 200 mg de INVIRASE® administradas como dosis única de 600 mg en 71 voluntarios sanos. En el estudio comparativo de bioequivalencia con los comprimidos de 500 mg y las cápsulas de 200 mg de INVIRASE® en combinación con ritonavir, se observó una diferencia en función del género: las mujeres presentaron exposiciones al saquinavir mayores que las de los hombres (ABC: 56%; Cmáx: 26%). No se observaron indicios de que la edad o el peso corporal pudieran explicar esta diferencia en función del género en este estudio. Con la dosis aprobada, no se han notificado diferencias clínicamente significativas de seguridad y eficacia entre los hombres y las mujeres. El tratamiento de pacientes masculinos y femeninos con 1.000 mg/100 mg de saquinavir/ritonavir dos veces al día se considera seguro y eficaz. Raza: no se ha determinado la influencia de la raza en la farmacocinética de INVIRASE®. No se han estudiado las características farmacocinéticas de INVIRASE® en pacientes geriátricos ( > 65 años) ni en niños ( < 16 años) (ver Precauciones generales). Pediátrico: existe información disponible acerca de la farmacocinética en estado estable de pacientes infectados con VIH en dos estudios de dosis múltiples. En el estudio NV20911, 5 pacientes eran < 2 años y 13 de entre 2 a < 6 años, y recibieron 50 mg/kg de saquinavir dos veces al día (sin exceder 1.000 mg bid) potenciado con ritonavir a 3 mg/kg en pacientes con un peso corporal que varió de 5 a < 15 kg o 2,5 mg/kg para pacientes con un peso corporal que varió de 15 a 40 kg (sin exceder 100 mg dos veces al día). En el estudio HIVNAT 017, 19 pacientes de entre 5 a 13 años de edad, recibieron 50 mg/kg de saquinavir dos veces al día (sin exceder 1.000 mg dos veces por día) potenciado con ritonavir a 57,5 mg/m2 (o 2,3 mg/kg) dos veces por día. Los parámetros de exposición farmacocinética de estos grupos se enlistan en la Tabla 6 que se encuentra abajo: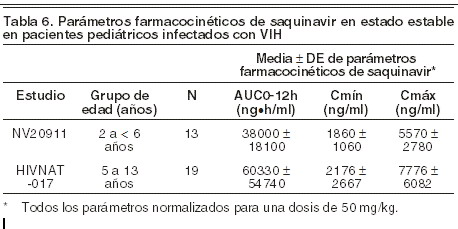
Se ha demostrado la farmacocinética, seguridad y actividad de saquinavir en 75 niños de 2 a 16 años tratados con INVIRASE® potenciado con ritonavir o INVIRASE® potenciado coformulado con lopinavir/ritonavir. Los datos de tres estudios en niños, PACTG 397, HIVNAT 017 (ML19540), y NV20911, demostraron que saquinavir potenciado con ritonavir a dosis bajas, proporcionó niveles plasmáticos de saquinavir que fueron más altos en comparación con saquinavir solo. PACTG 397 fue un estudio aleatorizado diseñado para evaluar la seguridad, tolerabilidad, biodisponibilidad, y actividad de las cápsulas de gelatina blanda de saquinavir solas y en combinación con ritonavir o nelfinavir en niños de 3 a 16 años de edad. Catorce pacientes fueron dosificados con saquinavir 50 mg/kg bid y ritonavir 100 mg bid con uno o dos inhibidores nucleósidos de la transcriptasa reversa (NRTIs) como tratamiento de base. Los pacientes fueron monitoreados en cuanto a seguridad y actividad durante 48 semanas de seguimiento. Las cápsulas de gelatina blanda de saquinavir administradas en combinación con ritonavir y uno o dos NRTIs fueron bien toleradas. HIVNAT 017 (ML19540) fue un estudio abierto, de un solo grupo, en dos centros diferentes en Tailandia, que involucró 50 niños de entre 4 y 15 años de edad, que evaluó la farmacocinética, seguridad, y actividad de lopinavir/ritonavir (230/57,5 mg/m2 bid) con saquinavir (50 mg/kg dos veces al día administrado como una cápsula de gel rígido de 200 mg) durante 96 semanas. Los primeros 20 niños reclutados fueron sometidos a un muestreo farmacocinético intensivo en estado estable para niveles plasmáticos de lopinavir, ritonavir, y saquinavir. El tratamiento con lopinavir/ritonavir 230/57,5 mg/m2 y saquinavir 50 mg/kg bid en niños de 4 a 16 años de edad, resultó en un beneficio del tratamiento durante 96 semanas, con una supresión sustancial de la carga viral (el porcentaje de pacientes con ARN de VIH < 400 copias/ml en la semana 96 fue de 56% para los faltantes = fracasos y de 93% para el análisis con tratamiento. El porcentaje de pacientes con ARN de VIH < 50 copias/ml en la semana 96, fue de 52% para los faltantes = fracasos y de 84% para el análisis con tratamiento), y la media de aumento del porcentaje de conteo de linfocitos CD4 aumentó de 7,6% en el tamizaje a 23,6% en la semana 96. El tratamiento fue bien tolerado sin eventos adversos nuevos o inesperados. NV20911 fue un estudio abierto, multicéntrico (Argentina, España y Tailandia) en 18 niños de 4 meses a menos de 6 años de edad que evaluó la farmacocinética, seguridad y actividad de saquinavir (50 mg/kg bid hasta la dosis adulta de 1.000 mg dos veces al día) y de la solución oral de ritonavir (3 mg/kg bid para un peso corporal de 5 a < 15 kg, 2,5 mg/kg dos veces al día para un peso corporal de 15 a 40 kg y 100 mg bid para un peso corporal > 40 kg más ≥2 ARVs de base). Los lactantes y niños pequeños fueron estratificados en 2 grupos: el Grupo A "Grupo de Edad Baja" de 4 meses a menos de 2 años de edad, y Grupo B "Grupo de Edad Alta" de niños de 2 años a menos de 6 años de edad. El tratamiento con saquinavir/ritonavir resultó en un estado virológico (porcentaje de pacientes con carga viral < 400 copias/ml en la semana 48 de 72% para faltantes = fracasos a 81% en el análisis con tratamiento. Los pacientes con carga viral < 50 copias/ml variaron de 61% faltantes = fracasos a 69% en el análisis con tratamiento después de 48 semanas de tratamiento), estado inmunológico (conteo de linfocitos CD4 expresado como porcentaje de aumento de media de CD4 de 2,9%), y beneficio clínico durante el período del estudio de 48 semanas. El tratamiento se consideró seguro y bien tolerado, y no se observaron eventos adversos nuevos o inesperados. Todos los eventos adversos reportados fueron leves o moderados en intensidad, y los eventos individuales reportados con mayor frecuencia fueron bronquitis, diarrea y vómito. En lactantes y niños que no pueden tragar las cápsulas de INVIRASE®, se mezcló el contenido de las cápsulas de 200 mg de INVIRASE® con jarabe dulce, o jarabe de sorbitol (en niños con diabetes Tipo I o intolerancia a la glucosa), o mermelada o fórmula para bebés (ver Dosis y vía de administración).
Contraindicaciones: INVIRASE® reforzado con ritonavir está contraindicado en pacientes con hipersensibilidad a saquinavir, ritonavir o a cualquier otro componente de los comprimidos. INVIRASE® reforzado no debe administrarse junto con fármacos con los que podría interactuar y provocar efectos secundarios potencialmente mortales. En la tabla 7 se presentan los fármacos que no deben administrarse con INVIRASE® reforzado (ver Interacciones medicamentosas y de otro género). INVIRASE® reforzado con ritonavir está contraindicado en pacientes con prolongación congénita o adquirida documentada - del QT, y alteraciones en los metabolitos particularmente la hipocalemia no corregida. INVIRASE® reforzado con ritonavir está contraindicado con algunos medicamentos que tienen ambas interacciones farmacocinéticas y prolongan el intervalo QT y/o PR (ver Interacciones medicamentosas y de otro género).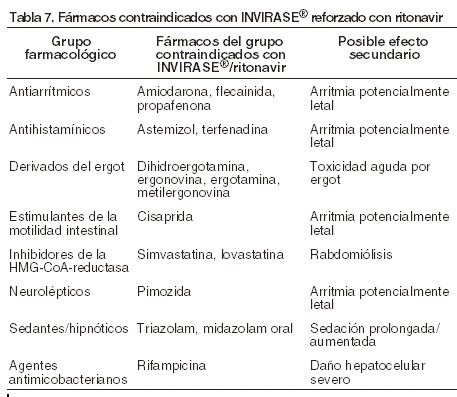
INVIRASE® reforzado con ritonavir está contraindicado en pacientes con insuficiencia hepática grave (ver Precauciones generales).
Precauciones.
generales: Aspectos que deben considerarse al empezar el tratamiento con INVIRASE®: INVIRASE® no debe administrarse sin ritonavir (no reforzado). INVIRASE® siempre debe administrarse en asociación con ritonavir (reforzado) (ver Dosis y vía de administración). Los pacientes deben saber que el saquinavir no cura la infección por el VIH, de modo que se pueden seguir presentando enfermedades asociadas a la infección avanzada por el VIH, como las infecciones oportunistas. Los pacientes también deben saber que podrían experimentar efectos no deseados asociados a la medicación administrada en forma simultánea. Insuficiencia hepática: no es necesario realizar ningún ajuste previo de la dosis para pacientes infectados por HIV con daño hepático moderado (ver Instrucciones de dosificación en poblaciones especiales). En pacientes con hepatitis B o C subyacente, cirrosis, alcoholismo crónico u otras alteraciones hepáticas, se han descrito casos de empeoramiento de la hepatopatía y aparición de hipertensión portal tras comenzar el tratamiento con saquinavir. Los síntomas asociados incluyen: ictericia, ascitis, edema y, en ocasiones, várices esofágicas. Varios de estos pacientes fallecieron. No se ha determinado una relación causal entre el tratamiento con saquinavir y la aparición de hipertensión portal (ver Contraindicaciones). Debe considerarse la conveniencia de aumentar la vigilancia de signos y síntomas de hepatotoxicidad. Insuficiencia renal: la depuración renal es la vía de eliminación menos importante, la principal vía de metabolismo y excreción para saquinavir es la hepática. Por lo tanto no se requiere ajustar la dosis inicial en los pacientes con insuficiencia renal (ver Instrucciones especiales de dosificación e Insuficiencia renal). Ahora bien, ya que no se han realizado estudios en pacientes con insuficiencia renal grave, se recomienda tener precaución en tales casos. Niños y ancianos: no se han determinado la seguridad y la eficacia de saquinavir en los niños y adolescentes menores de 16 años infectados por el VIH. La información sobre la cápsula de gelatina blanda de saquinavir no reforzado en niños es limitada y no existe en niños tratados con INVIRASE® no reforzado. Debido a que los niveles plasmáticos de saquinavir son significativamente inferiores en los niños en comparación con los adultos, INVIRASE® no reforzado no debe utilizarse en niños. La combinación de la cápsula de gelatina blanda de saquinavir (50 mg/kg dos veces al día) administrado con nelfinavir o ritonavir en niños incrementa considerablemente la exposición al saquinavir y cuando es combinado con ritonavir, la exposición a saquinavir podría superar más de dos veces la obtenida en adultos con 1.200 mg de la cápsula de gelatina blanda de saquinavir tres veces al día. La experiencia con personas mayores de 60 años es limitada. Intolerancia a la lactosa: cada cápsula contiene 63,3 mg de lactosa (anhidra) y cada comprimido contiene 38,5 mg de lactosa (monohidrato). Pacientes con problemas hereditarios de intolerancia a la galactosa, deficiencia de lactasa de los lapones (Lapp lactasa) o malabsorción de glucosa-galactosa (trastorno autosómico recesivo) no deben tomar estos medicamentos. Hemofilia: se ha descrito un aumento de las hemorragias (por ejemplo, hematomas cutáneos espontáneos y hemartrosis) en hemofílicos de tipos A y B tratados con inhibidores de la proteasa. A algunos pacientes se les administró factor VIII. En más de la mitad de los casos notificados, el tratamiento con los inhibidores de la proteasa no se suspendió o pudo reiniciarse tras una interrupción pasajera. Se ha invocado una posible relación causal, aunque todavía se desconoce cuál pueda ser el mecanismo de acción. Así pues, se debe informar a los hemofílicos sobre la posibilidad de que aumenten las hemorragias. Diabetes mellitus e hiperglucemia: se han descrito casos de diabetes mellitus de aparición reciente, hiperglucemia o reagudización de una diabetes preexistente en pacientes tratados con inhibidores de la proteasa. En algunos de ellos, la hiperglucemia fue grave y en ocasiones se asoció a cetoacidosis. Muchos pacientes presentaban condiciones médicas confusas, algunas de las cuales requerían tratamiento con fármacos que se han asociado al desarrollo de diabetes mellitus o de hiperglucemia. No se ha establecido una relación causal entre el tratamiento con inhibidores de la proteasa y la aparición de hiperglucemia o diabetes mellitus. Redistribución del tejido adiposo: en los pacientes que reciben terapia antirretroviral combinada, se ha observado redistribución y acumulación del tejido adiposo corporal, consistente en obesidad central, aumento del tejido adiposo dorsocervical (joroba de búfalo), emaciación periférica, hipertrofia de las mamas y "aspecto cushingoide". También se ha asociado a anomalías metabólicas, como hipertrigliceridemia, hipercolesterolemia, resistencia a la insulina e hiperglucemia. La gravedad de estos trastornos metabólicos varía dentro de cada uno de los tres grupos de antirretrovirales y entre ellos (inhibidores de la proteasa, inhibidores de la transcriptasa reversa análogos nucleósidos [ITRAN] e inhibidores de la transcriptasa reversa análogos no nucleósidos [ITRNN]). El mayor riesgo de lipodistrofia se ha asociado a edad avanzada, una larga duración del tratamiento antirretroviral, el uso de estavudina, la hipertrigliceridemia e hiperlactemia. La exploración clínica debe incluir la evaluación de los signos físicos de redistribución del tejido adiposo. Se aconseja vigilar la lipidemia y la glucemia. En caso de alteraciones metabólicas, debe considerarse la posibilidad de cambiar el tratamiento antirretroviral o administrar un tratamiento específico corrector de las mismas (por ejemplo, con hipolipemiantes). En la actualidad, se desconocen los mecanismos de estos fenómenos y de las consecuencias a largo plazo, incluido el aumento del riesgo de enfermedades cardiovasculares. Efectos sobre la capacidad para conducir y utilizar las máquinas: no se han realizado estudios sobre la capacidad para conducir vehículos y utilizar máquinas durante el tratamiento con INVIRASE®. No existen pruebas de que INVIRASE® pueda alterar la capacidad del paciente para conducir vehículos o manejar máquinas, sin embargo deben tenerse en cuenta los eventos adversos de INVIRASE® (ver Reacciones secundarias y adversas).
Restricciones de uso durante el embarazo y la lactancia: De los estudios realizados con animales de experimentación no se desprende ningún efecto nocivo, directo o indirecto, sobre el desarrollo embrionario o fetal, la evolución del embarazo o el desarr