KETEK®
SANOFI AVENTIS
Denominación genérica: Telitromicina.
Forma farmacéutica y formulación: Cada gragea contiene: telitromicina 400 mg. Excipiente cbp 1 gragea.
Indicaciones terapéuticas: KETEK® está indicado para el tratamiento de las siguientes infecciones en pacientes a partir de los 18 años de edad: neumonía adquirida en la comunidad, leve o moderada. KETEK® está indicado en: exacerbación aguda de bronquitis crónica. Sinusitis aguda. Cuando se trate de infecciones causadas por cepas que se sospeche o se conozca que son resistentes a beta-lactámicos y/o macrólidos, y que estén cubiertas por el espectro antibacteriano de telitromicina. Amigdalitis-faringitis debida a S. pyogenes, como una alternativa adecuada a los antibióticos beta-lactámicos, debido a que existe una alta prevalencia de S. pyogenes resistente a macrólidos, mediado por ermTR o mefA. Al prescribir telitromicina, se deberán considerar los lineamientos para el uso apropiado de medicamentos antibacterianos y la prevalencia local de resistencia.
Farmacocinética y farmacodinamia: La telitromicina es el primer miembro de una nueva familia de antibióticos, llamada ketólidos, que se relacionan con el grupo de antibióticos MLSB (macrólidos, lincosamidas y estreptograminas B). Farmacodinamia: la telitromicina inhibe la síntesis proteica bacteriana al bloquear la traducción del RNA ribosomal (RNAr) 23S bacteriano de la subunidad 50S, e inhibir la unión de subunidades ribosomales nacientes. La telitromicina bloquea la síntesis proteica al unirse a dos sitios de la subunidad ribosomal 50S: los dominios II y V del RNAr 23S. Se ha determinado que la afinidad de telitromicina por el RNAr 23S es 10 veces mayor que la de eritromicina A en cepas sensibles a eritromicina, y 25 veces mayor en cepas resistentes a macrólidos. La diferencia en la fortaleza de unión puede atribuirse a la cadena lateral de carbamato en C11-12. Esto permite que telitromicina se mantenga unida al dominio II, aun en presencia de resistencia que altera el sitio de unión del dominio V. Al mantener la unión en el dominio II, telitromicina retiene actividad contra la mayoría de los cocos grampositivos (p. ej., S. pneumoniae) que expresan al gene (erm) de metilasa. Espectro antibacteriano: la telitromicina tiene potente actividad contra cocos grampositivos como Streptococcus pneumoniae, Streptococcus pyogenes y Staphylococcus aureus, cocos gramnegativos como Moraxella catarrhalis, y algunos bacilos gramnegativos como Haemophilus influenzae y Bordetella pertussis. Es altamente concentrada en fagocitos y posee buena actividad antibacteriana contra microorganismos patógenos respiratorios intracelulares y atípicos como Chlamydophila (Chlamydia) pneumoniae, Legionella pneumophila y microorganismos atípicos como Mycoplasma pneumoniae. La telitromicina muestra actividad bactericida in vitro contra Streptococcus pneumoniae (inclusive las cepas resistentes a eritromicina A y a penicilina G), Chlamydophila (Chlamydia) pneumoniae, Streptococcus pyogenes, Staphylococcus aureus, Haemophilus influenzae y Legionella pneumophila. La actividad de telitromicina contra S. pneumoniae es independiente de la sensibilidad de las cepas aisladas a otras clases de antibióticos, p. ej., penicilinas, cefalosporinas, macrólidos, cotrimoxazol, tetraciclinas y fluoroquinolonas. La telitromicina también mantiene elevada actividad contra S. pneumoniae con un mecanismo subyacente de resistencia a eritromicina A. Puntos de corte: la prevalencia de resistencia puede variar geográficamente y con el tiempo para ciertas especies, por lo que es deseable la información local sobre resistencia, particularmente cuando se trata de infecciones graves. La información de la siguiente tabla proporciona únicamente una guía aproximada sobre la sensibilidad probable de los microorganismos a telitromicina. Los puntos de corte de la CMI recomendados para telitromicina, separando a los microorganismos sensibles de los medianamente sensibles, y a los microorganismos medianamente sensibles de los resistentes, son: S. pneumoniae y otros: sensibles ≤1 mg/l; resistentes ≥4 mg/l. H. influenzae: sensibles ≤2 mg/l; resistentes ≥8 mg/l.
Resistencia: la telitromicina no induce resistencia MLSB in vitro en cepas de S. aureus, S. pneumoniae y S. pyogenes, un atributo relacionado a su función 3-ceto. Se ha demostrado in vitro que rara vez se presenta resistencia a telitromicina debida a mutación espontánea. Efecto sobre la flora oral y fecal: en un estudio comparativo realizado en voluntarios sanos, telitromicina en dosis de 800 mg diarios y claritromicina en dosis de 500 mg dos veces al día durante 10 días, redujeron de manera semejante y reversible la flora oral y fecal. Sin embargo, ninguna cepa resistente de estreptococos del grupo viridans emergió en saliva de pacientes en tratamiento con telitromicina, a diferencia de lo que sucedió con claritromicina. Farmacocinética: absorción: la absorción de telitromicina es rápida después de su administración por vía oral. La biodisponibilidad absoluta es de 57% tanto en sujetos jóvenes como de edad avanzada, después de administrar una sola dosis de 800 mg. La proporción y el grado de absorción no se ven afectados por la ingestión de alimentos, por lo que las grageas de KETEK® pueden ser administradas con los alimentos. En sujetos sanos, las concentraciones plasmáticas máximas de aproximadamente 2 mg/l se alcanzan dentro de una mediana de 1 hora, después de administrar una dosis de 800 mg de telitromicina. Las concentraciones plasmáticas mínimas en el estado estacionario son alcanzadas dentro de 2 a 3 días, al administrar una dosis diaria de 800 mg de telitromicina. El ABC en el estado estacionario se incrementa aproximadamente 1,5 veces, comparada con la dosis única. La vida media de eliminación terminal promedio es 10 horas. La farmacocinética de telitromicina después de la administración de una y múltiples (7 días) dosis de 800 mg una vez al día a sujetos adultos sanos, se muestra en la siguiente tabla: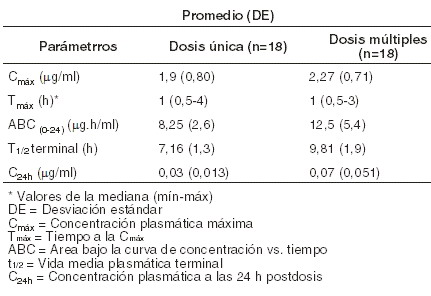
En una población de pacientes, las concentraciones plasmáticas máxima y mínima promedio en el estado estacionario fueron 2,9 mg/ml (± 1,55), (n=219) y 0,2 mg/ml (± 0,22), (n=204), respectivamente, después de 3 a 5 días de la administración de 800 mg de telitromicina una vez al día. Distribución: la unión total a proteínas in vitro es de aproximadamente 60% a 70%, y es debida sobre todo a la albúmina sérica humana. La unión a proteínas no es modificada en sujetos de edad avanzada ni en pacientes con insuficiencia hepática. El volumen de distribución de telitromicina es 2,9 l/kg, después de la perfusión intravenosa. Las concentraciones de telitromicina observadas en los tejidos de las vías respiratorias, leucocitos y macrófagos alveolares son más elevadas que la CMI para telitromicina, contra los microorganismos patógenos de las vías respiratorias indicados. Después de su administración, el fármaco persistió 48 horas en el líquido de revestimiento epitelial y en los macrófagos alveolares. Las concentraciones de telitromicina en mucosa bronquial, líquido de revestimiento epitelial, macrófagos alveolares y amígdalas, después de la administración de 800 mg una vez al día durante 5 días, se muestran en la siguiente tabla: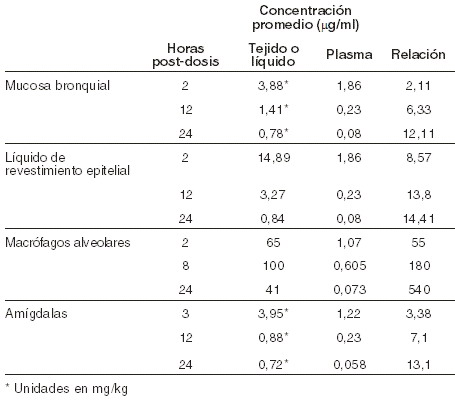
La telitromicina es altamente concentrada por leucocitos y es eliminada más lentamente de leucocitos que del plasma. Las concentraciones promedio de telitromicina en leucocitos alcanzaron un pico en 72,1 mg/ml a las 6 horas, y se mantuvieron en 14,1 mg/ml 24 horas después de 5 días de la administración repetida de 600 mg una vez al día. Después de 10 días de la administración repetida de 600 mg una vez al día, las concentraciones en leucocitos permanecieron en 8,9 mg/ml 48 horas después de la última dosis. Se presentaron altas concentraciones en leucocitos y macrófagos alveolares, lo que puede contribuir a la distribución del fármaco en los tejidos inflamados: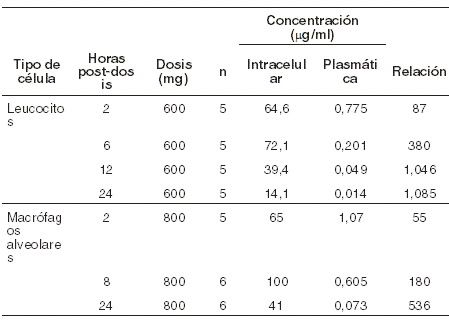
Biotransformación: la telitromicina se metaboliza principalmente en hígado. El principal compuesto que circula en el plasma después de la administración de una dosis de 800 mg marcada radioactivamente, fue el compuesto de origen, que representó el 56,7% del ABC total de telitromicina. El metabolito principal representó 12,6% del ABC de telitromicina. Se cuantificaron otros tres metabolitos plasmáticos, cada uno de los cuales representó el 3% o menos del ABC de telitromicina. Telitromicina es metabolizada por isoenzimas del citocromo P450 (CYP450) y por vías independientes de CYP450. Eliminación: después de la administración de telitromicina marcada radioactivamente, 76% de la radioactividad fue recuperada de las heces, con un 17% adicional recuperado de la orina. Aproximadamente una tercera parte de telitromicina fue eliminada sin cambios; 20% en heces y 12% en orina. La depuración total es aproximadamente 70 l/h, correspondiendo el 17% a la depuración renal. Poblaciones especiales: género: no se observaron diferencias significativas en la farmacocinética de telitromicina entre hombres y mujeres. Insuficiencia hepática: en un estudio de dosis única (800 mg) en 12 pacientes y en un estudio de dosis múltiples (800 mg) en 13 pacientes con insuficiencia hepática ligera a grave (grado A, B y C de Child-Pugh), la Cmáx, el ABC y el t1/2 de telitromicina fueron similares en comparación con aquellas obtenidas en sujetos sanos de igual género y edad. En ambos estudios fue observado un aumento en la eliminación renal en pacientes con insuficiencia hepática, lo que indica que esta vía puede compensar parcialmente la disminución en la depuración metabólica. No se requiere ajuste de la dosis debido a insuficiencia hepática (ver Dosis y vía de administración). Insuficiencia renal: en un estudio de dosis múltiples, 36 sujetos con grados variables de insuficiencia renal recibieron ya sea 400 mg, 600 mg u 800 mg de KETEK® una vez al día durante 5 días. Las farmacocinéticas plasmáticas de telitromicina en el estado estacionario, a las dosis de 400 y 600 mg, fueron similares a través de los diferentes grupos con deterioro renal. Hubo un aumento de 1,4 veces en la Cmáx en el estado estacionario, y de 1,9 veces en el ABC (0-24) en el estado estacionario, con dosis múltiples de 800 mg, dentro del grupo con deterioro renal grave (depuración de creatinina < 30 ml/min), en comparación con voluntarios sanos. La excreción renal puede servir como una vía de eliminación compensatoria para telitromicina, en situaciones donde está deteriorada la depuración metabólica. Los pacientes con deterioro renal grave están propensos a condiciones que pueden deteriorar su depuración metabólica. Por tanto, en presencia de insuficiencia renal grave (depuración de creatinina < 30 ml/min), se recomienda reducción de la dosis de KETEK® (ver Dosis y vía de administración). En un estudio de dosis única, en pacientes con insuficiencia renal terminal hemodializados (n=10), los valores promedio de Cmáx y de ABC fueron similares a los de sujetos sanos, cuando KETEK® fue administrado dos horas después de la diálisis. Insuficiencia múltiple: los efectos del deterioro de múltiples vías de eliminación fueron estudiados en 12 sujetos ≥60 años de edad, con función renal disminuida (depuración de creatinina =24 a 80 ml/min), y administrando ketoconazol para bloquear la vía del citocromo P450 3A4 (CYP3A4). Cuando en este estudio estuvieron presentes la insuficiencia renal grave (depuración de creatinina < 30 ml/min) y el deterioro concomitante del metabolismo hepático, la exposición a la telitromicina estuvo en el extremo más alto de los valores observados en los estudios clínicos fase III. En presencia de insuficiencia renal grave (depuración de creatinina < 30 ml/min), con o sin insuficiencia hepática coexistente, se recomienda reducción de la dosis de KETEK® (ver Dosis y vía de administración). Sujetos de edad avanzada: en pacientes con infecciones respiratorias, de más de 65 años de edad (n=20), la Cmáx y el ABC de telitromicina plasmática presentaron un aumento del 30% y 40%, respectivamente, en comparación con pacientes de menos de 65 años de edad (n=142). En sujetos de entre 65 a 92 años de edad (n=14), la Cmáx y el ABC de telitromicina plasmática presentaron un aumento del 100%, después de administrar 800 mg una vez al día durante 10 días, en comparación con adultos sanos de entre 19 a 29 años de edad (n=12). Durante los estudios fase III, los sujetos de edad avanzada recibieron la misma dosis de telitromicina (800 mg) que los sujetos más jóvenes, sin que se advirtieran diferencias en la seguridad y la eficacia entre los grupos. Pacientes pediátricos: la farmacocinética de telitromicina en pacientes adolescentes, de entre 13 a 17 años de edad (n=18), fue semejante a la observada en voluntarios adultos sanos. No ha sido estudiada la farmacocinética de telitromicina en poblaciones pediátricas de 12 años de edad e inferiores.
Contraindicaciones: Antecedentes de hipersensibilidad a telitromicina y/o a alguno de los componentes de la fórmula, o a algún macrólido. Administración concomitante de KETEK® con alguno de los siguientes fármacos: cisaprida, pimozida, astemizol y terfenadina. KETEK® no deberá ser administrado en pacientes con antecedentes de hepatitis y/o ictericia asociadas al uso de telitromicina. KETEK® está contraindicado en pacientes con miastenia gravis. Se han reportado exacerbaciones de miastenia gravis, las cuales en algunas ocasiones han ocurrido dentro de las primeras horas después de la administración de la primera dosis de telitromicina. Los reportes han incluido muerte e insuficiencia respiratoria aguda de inicio rápido que pone en peligro la vida.
Precauciones generales: Como con casi todos los fármacos antibacterianos, la diarrea, particularmente si es grave, persistente y/o sanguinolenta, que se presenta durante o después del tratamiento con telitromicina puede ser sintomática de la colitis pseudomembranosa. Si se sospecha de colitis pseudomembranosa, KETEK® debe ser suspendido inmediatamente y los pacientes deben ser tratados con terapia específica y/o medidas de soporte. Existe la posibilidad de que telitromicina prolongue el intervalo QTc del electrocardiograma en algunos pacientes. La prolongación de QTc puede inducir a un mayor riesgo de arritmias ventriculares, inclusive torsades de pointes (TdP). Por lo tanto, KETEK® debe ser evitado en pacientes con prolongación congénita del intervalo QTc, con hipokaliemia incorregible (≤3 mmol/l [mEq/l]), hipomagnesemia, bradicardia ( < 50 bpm), y/o en pacientes a los que se administran fármacos antiarrítmicos Clase IA (p. ej., quinidina y procainamida) o Clase III (p. ej., dofetilida). En los estudios clínicos, el efecto sobre QTc fue reducido (promedio de aproximadamente 1 mseg). En estudios comparativos, se observaron efectos similares a los de claritromicina con un ∆QTc > 30 mseg durante el tratamiento, en el 7,6% y 7,0% de los casos, respectivamente. Ningún paciente de cualquiera de los dos grupos presentó un ∆QTc > 60 mseg. No se informó de TdP u otras arritmias ventriculares graves ni de sincope relacionado, en el programa clínico, y no se identificaron subgrupos de riesgo. La telitromicina puede causar efectos adversos tales como trastornos visuales ( < 1%), que pueden reducir la capacidad para realizar ciertas tareas. KETEK® puede causar trastornos visuales particularmente al reducir la capacidad de acomodación y al reducir la capacidad de liberación de la acomodación. Las alteraciones visuales incluyeron visión borrosa, dificultad para enfocar y diplopía. La mayoría de los eventos fueron leves a moderados; sin embargo, se han reportado casos severos. Se han reportado casos de eventos adversos post-comercialización relacionados a pérdida transitoria de la conciencia incluyendo algunos casos asociados a síndrome vagal. A causa de las potenciales alteraciones visuales o pérdida de la conciencia, los pacientes deberán tratar de minimizar actividades tales como conducir vehículos, operar maquinaria o algún otro tipo de actividad riesgosa durante el tratamiento con KETEK®. Si los pacientes experimentan alteraciones visuales o pérdida de la conciencia al encontrarse bajo tratamiento con KETEK®, no deberán conducir vehículos, operar maquinaria o realizar actividades riesgosas. Se ha observado de manera común la alteración de enzimas hepáticas en estudios clínicos con telitromicina. Se han reportado casos post-comercialización de hepatitis severa e insuficiencia hepática, en algunos casos fatal, los cuales generalmente han estado asociados a enfermedades subyacentes serias o medicamentos concomitantes. Estas reacciones hepáticas fueron observadas durante o poco después del tratamiento, y en la mayoría de los casos fueron reversibles después de la suspensión de la telitromicina. Médicos y pacientes deben vigilar la aparición de signos y síntomas de enfermedad hepática tales como anorexia, ictericia, orina de color oscuro, prurito o dolor abdominal. En caso de signos y síntomas de hepatitis, se deberá indicar a los pacientes suspender el tratamiento y contactar a su médico.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: no hay datos sobre el uso de KETEK® en mujeres embarazadas. En dosis superiores a 150 mg/kg y 20 mg/kg en ratas y conejos, respectivamente, la toxicidad materna dio como resultado retardo de la maduración fetal. KETEK® no debe usarse durante el embarazo a menos que los beneficios esperados para la paciente superen al posible riesgo para el feto. Lactancia: la telitromicina es excretada en la leche de animales que amamantan. No se dispone de datos correspondientes para humanos. KETEK® no debe usarse durante la lactancia a menos que los beneficios esperados para la paciente superen al posible riesgo para el neonato.
Reacciones secundarias y adversas: Se ha utilizado la siguiente clasificación de frecuencias de aparición de efectos adversos: muy común = ≥10%. Común = 1-10% de los pacientes. Poco común = 0,1-1% de los pacientes. Raro = 0,01-0,1% de los pacientes. Muy raro = 0,01% o menos de los pacientes. Sistema gastrointestinal: muy común: diarrea. Común: náusea, vómito, dolor gastrointestinal y flatulencia. Poco común: estreñimiento, anorexia, candidiasis oral y estomatitis. Alergia: poco común: eritema, urticaria y prurito. Muy raro: reacciones alérgicas graves, inclusive angioedema y anafilaxia. Sistema biliar y hepático: común: aumento de enzimas hepáticas (AST, ALT, fosfatasa alcalina). Poco común: hepatitis, la mayoría de los reportes de disfunción hepática fueron leves a moderados. Raro: ictericia colestásica. Sistema nervioso: común: mareo y cefalea. Poco común: somnolencia, insomnio y nerviosismo, vértigo. Raro: parestesia. Sistema linfático y hemático: poco común: eosinofilia. Organos sensoriales: común: trastornos del gusto. Poco común: ha habido reportes ( < 1%) de trastornos visuales asociados con el uso de KETEK®, incluyendo visión borrosa, dificultad para enfocar y diplopía. La mayoría de los casos fueron leves a moderados. Por lo general, los incidentes visuales ocurrieron unas cuantas horas después de la administración de la primera o segunda dosis, se repitieron al administrar dosis posteriores, permanecieron varias horas y fueron completamente reversibles tanto durante la terapia como después de finalizado el tratamiento. Estos casos no se han asociado con signos de anormalidades oculares (ver Precauciones generales). Sistema urogenital: común: candidiasis vaginal. Piel: raro: eccema. Musculoesquelético: raro: calambres musculares, exacerbación de miastenia gravis (ver Contraindicaciones). Trastornos cardiovasculares: poco común: rubor. Raro: arritmia atrial, hipotensión y bradicardia. Muy raro: palpitaciones. Adicionalmente, los siguientes efectos adversos se han reportado: prolongación del intervalo QT/QTc, insuficiencia hepática severa (que generalmente se ha asociado con enfermedades subyacentes serias o uso de medicamentos concomitantes), hipersensibilidad, eritema multiforme, edema facial, pancreatitis, pérdida transitoria de la conciencia/síncope la cual puede estar precedida por síntomas vagales (ver Precauciones generales)
Interacciones medicamentosas y de otro género: Alimentos: no hay interacción con los alimentos, por lo que las grageas de KETEK® pueden tomarse con o sin alimentos. Interacciones con medicamentos: telitromicina es un inhibidor de CYP3A4 in vitro. La administración concomitante de medicamentos que son metabolizados principalmente por esta enzima puede provocar aumento de las concentraciones plasmáticas, lo que puede resultar eventualmente en aumento de eventos adversos. Debe tenerse precaución durante la administración concomitante de otros medicamentos que son sustratos de CYP3A4 (ver Contraindicaciones y Precauciones generales). Telitromicina es un inhibidor ligero de CYP2D6. Telitromicina es metabolizada principalmente por el citocromo P450 3A4 (CYP3A4) y en menor grado por el citocromo P450 1A (CYP1A). En estudios de farmacología clínica se comprobaron las siguientes interacciones fármaco-fármaco: cisaprida: las concentraciones plasmáticas máximas en el estado estacionario de cisaprida (un fármaco capaz de aumentar el intervalo QT), fueron aumentadas 95% cuando se administró concomitantemente con dosis repetidas de telitromicina, lo que provocó aumentos significativos en el intervalo QTc. En consecuencia, la administración concomitante de telitromicina y cisaprida está contraindicada (ver Contraindicaciones). Digoxina: se ha demostrado que KETEK® aumenta las concentraciones plasmáticas de la digoxina. En voluntarios sanos, las concentraciones plasmáticas mínimas al estado estacionario se incrementaron 21%. Sin embargo, no se observaron cambios significativos en los parámetros del ECG ni signos de toxicidad por digoxina. Deberá considerarse el monitoreo de los efectos secundarios o de las concentraciones plasmáticas de digoxina durante la administración concomitante de digoxina y telitromicina. Estatinas: cuando la simvastatina fue coadministrada con KETEK®, hubo un incremento de 5,3 veces en la Cmáx y de 8,9 veces en el ABC de la simvastatina, así como un aumento de 15 veces en la Cmáx y de 12 veces en el ABC de la simvastatina ácida. En otro estudio, cuando la simvastatina y la telitromicina se administraron separadas por 12 horas, hubo un incremento de 3,4 veces en la Cmáx y de 4,0 veces en el ABC de la simvastatina, así como un incremento de 3,2 veces en la Cmáx y de 4,3 veces en el ABC del metabolito activo. La interacción observada es, en promedio, del mismo orden de magnitud que la observada con eritromicina. El riesgo de miopatía se puede aumentar con altas concentraciones de simvastatina. Por lo tanto, debe ser evitada la administración concomitante de KETEK® con simvastatina u otras estatinas metabolizadas sobre todo por CYP3A4. Si se prescribe KETEK®, se puede considerar ya sea suspender la terapia de estas estatinas por la duración del tratamiento, o separar la administración de ambos productos por 12 horas. Con base a los resultados de este estudio, a las propiedades farmacocinéticas de las otras estatinas y a las interacciones reportadas para las otras estatinas debido a la inhibición de CYP3A4, cabe esperar que telitromicina produzca una interacción similar con lovastatina y una interacción menor con atorvastatina. Por lo que se sabe, pravastatina y fluvastatina no son metabolizadas por CYP3A4; en consecuencia, no se espera interacción alguna. Teofilina: cuando se coadministró KETEK®, no hubo efecto farmacocinético de relevancia clínica sobre la formulación de liberación prolongada de teofilina. Sin embargo, la coadministración de telitromicina y teofilina debe ser separada por 1 hora, con objeto de reducir la probabilidad de efectos adversos gastrointestinales. Itraconazol y ketoconazol: los estudios de interacción de dosis múltiples realizados en sujetos jóvenes con itraconazol y ketoconazol, dos inhibidores del CYP3A4, demostraron que las concentraciones plasmáticas máximas de telitromicina fueron incrementadas 22% y 51%, respectivamente, y el ABC 54% y 95%, respectivamente. La vida media terminal de telitromicina no tuvo cambios en presencia de itraconazol y ketoconazol. Estos cambios en la farmacocinética de telitromicina no requieren ajuste de dosis, ya que la exposición a telitromicina permanece dentro de un intervalo bien tolerado. Anticoagulantes orales/warfarina: no hubo efecto farmacodinámico o farmacocinético sobre la mezcla racémica de warfarina, cuando se coadministró con KETEK® en sujetos sanos. Los reportes espontáneos poscomercialización sugieren que la administración concomitante de KETEK® y de anticoagulantes orales puede potenciar los efectos de los anticoagulantes orales. Deberá considerarse el monitoreo de los tiempos de protrombina/INR mientras que los pacientes estén recibiendo simultáneamente KETEK® y anticoagulantes orales. Anticonceptivos orales: en un estudio de interacción farmacocinética/farmacodinámica, KETEK® no interfirió con el efecto anovulatorio de los anticonceptivos orales que contienen etinilestradiol y levonorgestrel. Ranitidina, antiácidos: la ranitidina o los antiácidos que contienen hidróxido de aluminio y de magnesio no evidenciaron interacción farmacocinética clínicamente relevante sobre telitromicina. Paroxetina: cuando se coadministró KETEK®, no hubo efecto farmacocinético sobre paroxetina, un sustrato de CYP2D6. Benzodiazepinas: la administración concomitante de telitromicina con midazolam intravenoso u oral, provocó aumentos de 2 y 6 veces, respectivamente, en el ABC de midazolam, debido a la inhibición del metabolismo de midazolam dependiente de CYP3A4. Los pacientes a los que se administre concomitantemente midazolam deberán ser monitoreados y, de ser necesario, deberá considerarse el ajuste de la dosis de midazolam. Deberá actuarse con precaución cuando se coadministren otras benzodiazepinas que son metabolizadas por CYP3A4 (p. ej., triazolam y en menor medida, alprazolam). Es improbable la interacción con telitromicina en el caso de las benzodiazepinas que no son metabolizadas por CYP3A4 (p. ej., temazepam, nitrazepam y lorazepam). Sotalol: se ha demostrado que KETEK® reduce la Cmáx de sotalol en un 34% y el ABC en un 20%, debido a una disminución de la absorción. Rifampicina: la Cmáx y el ABC de KETEK® fueron disminuidas 79% y 86%, respectivamente, durante la administración concomitante de rifampicina y KETEK® en dosis repetidas. Debe evitarse el tratamiento con KETEK® durante y 2 semanas después del tratamiento con inductores de CYP3A4 (tales como rifampicina, fenitoína, carbamazepina y el extracto de Hypericum perforatum [Hierba de San Juan]). Metoprolol: cuando el metoprolol fue coadministrado con KETEK®, hubo un aumento de aproximadamente 38% sobre la Cmáx y el ABC de metoprolol, sin efecto sobre la vida media de eliminación de metoprolol. La exposición de KETEK® no es modificada con la administración concomitante de dosis únicas de metoprolol. En los pacientes tratados con metoprolol por insuficiencia cardíaca, el incremento del metoprolol, un sustrato del CYP2D6, puede tener relevancia clínica. Por lo tanto, debe ser considerada con precaución la administración concomitante de KETEK® y metoprolol en pacientes con insuficiencia cardíaca. Las siguientes interacciones con otros medicamentos no se han estudiado con telitromicina, pero se han descrito con macrólidos: alcaloides derivados del cornezuelo de centeno (tales como ergotamina y dihidroergotamina). Se ha informado de vasoconstricción grave ("ergotismo") posiblemente con necrosis de las extremidades, cuando los antibióticos macrólidos fueron coadministrados con alcaloides derivados del cornezuelo de centeno. Sin datos adicionales, no se recomienda la administración concomitante de KETEK® y estos medicamentos. Pimozida, astemizol y terfenadina: se ha informado que los macrólidos alteran la biotransformación de estos medicamentos y aumentan sus concentraciones plasmáticas. Esto puede resultar en prolongación del intervalo QT y arritmias cardíacas, que incluyen taquicardia ventricular, fibrilación ventricular y torsades de pointes. Por consiguiente, está contraindicada la administración concomitante de telitromicina y cualquiera de estos medicamentos. Otras interacciones: con medicamentos metabolizados por el sistema del citocromo P450, tales como quinidina, carbamazepina, ciclosporina, hexobarbital, disopiramida y fenitoína es posible que se observe aumento de las concentraciones plasmáticas de los mismos cuando se coadministran con telitromicina.
Alteraciones en los resultados de pruebas de laboratorio: Aumento de enzimas hepáticas (AST, ALT, fosfatasa alcalina).
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Carcinogenicidad: no se han llevado a cabo estudios de largo plazo en animales, para determinar el potencial carcinogénico de KETEK®. Toxicidad reproductiva: la telitromicina no fue teratogénica en ratas ni en conejos. En dosis superiores a 150 mg/kg y 20 mg/kg en ratas y conejos, respectivamente, la toxicidad materna dio como resultado retardo de la maduración fetal. Una ligera reducción en los índices de fertilidad fue observada en ratas con dosis superiores a 150 mg/kg, tóxicas para las crías. Genotoxicidad: no se observó evidencia de genotoxicidad con telitromicina en los siguientes cuatro ensayos: mutación genética en células bacterianas, mutación genética en células de mamíferos, aberración cromosómica en linfocitos humanos y en la prueba del micronúcleo en el ratón. Otras toxicidades: en los estudios de toxicidad con dosis repetidas de telitromicina administradas durante 1, 3 y 6 meses en la rata, el perro y el mono, se demostró que el hígado fue el principal blanco de toxicidad, con aumento de enzimas hepáticas y evidencia histológica de daño. Estos efectos presentaron una tendencia a la regresión después del cese del tratamiento. Las exposiciones plasmáticas basadas en la fracción libre del fármaco, en los niveles sin observación de efectos adversos, oscilaron entre 1,6 a 13 veces la exposición clínica esperada. Se ha observado fosfolipidosis (acumulación intracelular de fosfolípidos) que afecta a varios órganos y tejidos (p. ej., hígado, riñón, pulmón, timo, bazo, vesícula biliar, nodos linfáticos mesentéricos, tracto gastrointestinal) en ratas y perros, a los que se administró telitromicina en dosis repetidas de 150 mg/kg/día o más durante un mes, y de 20 mg/kg/día o más durante 3-6 meses. Esta administración corresponde a concentraciones de exposición sistémica del fármaco libre de al menos 9 veces las concentraciones esperadas en humanos después de un mes, y de menos de una vez la concentración esperada en humanos después de 6 meses, respectivamente. Al suspender el tratamiento hubo evidencia de reversibilidad. Se desconoce el significado de estos hallazgos para el humano. Los estudios farmacológicos señalaron un efecto de prolongación tanto del intervalo QTc en perros, como de la duración del potencial de acción en fibras de Purkinje de conejo in vitro. Estos efectos se observaron con concentraciones de fármaco libre 8 a 13 veces las circulantes en el uso clínico.
Dosis y vía de administración: Dosis estándar y duración del tratamiento en adultos: en adultos y adolescentes a partir de los 13 años de edad, la dosis recomendada es de 800 mg una vez al día, es decir, dos grageas de 400 mg una vez al día. De acuerdo a la indicación, la duración del tratamiento será la siguiente: neumonía adquirida en la comunidad: 800 mg, una vez al día, durante 7 a 10 días. Exacerbación aguda de bronquitis crónica: 800 mg, una vez al día, durante 5 días. Sinusitis aguda: 800 mg, una vez al día, durante 5 días. Amigdalitis/faringitis: 800 mg, una vez al día, durante 5 días. Se deberá considerar el tomar KETEK® a la hora de acostarse para reducir el impacto potencial de alteraciones visuales y pérdida de la conciencia. Poblaciones especiales: pacientes pediátricos: no se ha establecido aún la eficacia y seguridad de telitromicina en poblaciones pediátricas de menos de 13 años de edad. Pacientes de edad avanzada: con base únicamente en la edad, no es necesario ajuste alguno de la dosis en pacientes de edad avanzada. Insuficiencia hepática: no es necesario ningún ajuste de la dosis en pacientes con insuficiencia hepática leve, moderada o severa, a menos que la función renal esté severamente deteriorada. Insuficiencia renal: no es necesario ningún ajuste de la dosis en pacientes con insuficiencia renal leve o moderada. En presencia de insuficiencia renal severa (depuración de creatinina < 30 ml/min), la dosis deberá reducirse a la mitad (es decir, 400 mg una vez al día). Hemodiálisis: en pacientes hemodializados, las grageas deberán administrase después de la sesión de diálisis, en los días en que se aplique este procedimiento. Administración: las grageas de KETEK® pueden ser administradas con o sin los alimentos.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: En caso de sobredosis aguda, debe realizarse vaciamiento gástrico ya sea induciendo vómito o efectuando lavado gástrico. Los pacientes deben ser observados cuidadosamente (p. ej., ECG, electrolitos), dando tratamiento sintomático y de soporte. Debe mantenerse una hidratación adecuada.
Presentación(es): Caja con 10, 14 y 20 grageas, en envase de burbuja.
Recomendaciones sobre almacenamiento: Consérvese a temperatura ambiente a no más de 30°C y en lugar seco.
Leyendas de protección: Su venta requiere receta médica. No se deje al alcance de los niños. Literatura exclusiva para médicos.
Nombre y domicilio del laboratorio: Sanofi-Aventis de México S.A. de C.V. Oficinas: Av. Universidad No. 1738, 04000, Coyoacán, México, D.F. Planta: Acueducto del Alto Lerma No. 2, Zona Industrial Ocoyoacac, 52740 Ocoyoacac, Edo. de México. ® Marca registrada.
Número de registro del medicamento: 372M2001 SSA IV.
Clave de IPPA: BEAR-04330020510071/RM 2005