KYPROLIS
AMGEN
Denominación genérica: Carfilzomib.
Forma farmacéutica y formulación: KYPROLIS es un polvo liofilizado blanco a blanquecino estéril para solución para inyección/mediante infusión. El frasco ámpula con polvo liofilizado contiene: Carfilzomib 60 mg Excipiente cbp.
Indicaciones terapéuticas: Mieloma Múltiple en Recaída o Refractario: KYPROLIS está indicado en combinación con dexametasona o con lenalidomida más dexametasona para el tratamiento de pacientes con mieloma múltiple en recaída o refractario que han recibido de una a tres líneas de terapia. KYPROLIS está indicado como agente único para el tratamiento de pacientes con mieloma múltiple en recaída o refractario, y que hayan recibido una o más líneas de terapia.
Farmacocinética y farmacodinamia: Farmacocinética: Carfilzomib a dosis entre 20 mg/m2 y 70 mg/m2 administrados en pacientes con mieloma múltiple mediante infusión de 30 minutos, dio lugar a aumentos en las concentraciones plasmáticas máximas (Cmáx) y el área bajo la curva con tiempo hasta el infinito (ABCinf) dependientes de la dosis. También se observó un aumento dependiente de la dosis en Cmáx y ABCinf entre dosis de carfilzomib de 20 mg/m2 y 56 mg/m2 mediante infusión de 2 a 10 minutos en pacientes con mieloma múltiple en recaída o refractaria. Una infusión de 30 minutos dio como resultado una ABCinf similar, pero con una Cmáx de 2 a 3 veces más baja que la observada mediante infusión de 2 a 10 minutos con la misma dosis. No hubo evidencia de acumulación de carfilzomib después de la administración repetida de 70 mg/m2 de carfilzomib mediante infusión semanal de 30 minutos o 15 mg/m2 y 20 mg/m2 mediante infusión de 2 a 10 minutos dos veces por semana.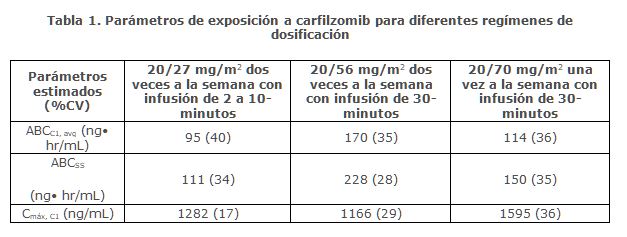
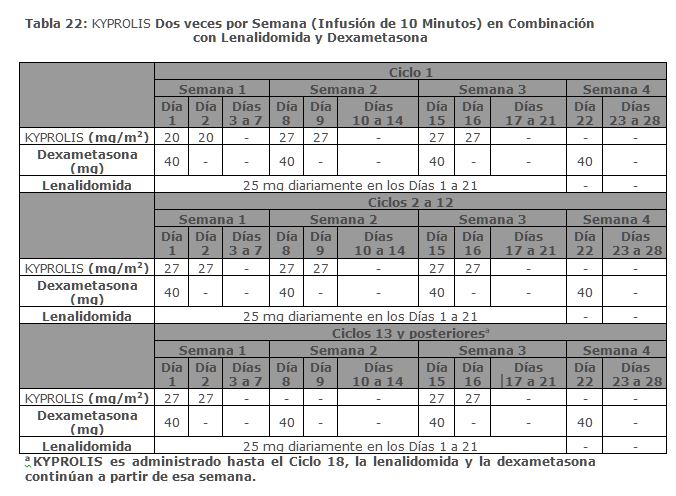
Distribución: El volumen de distribución promedio en estado estable de una dosis de carfilzomib de 20 mg/m2 fue de 28 L. Carfilzomib se une en un 97% a las proteínas plasmáticas humanas en el rango de concentraciones de 0. 4 micromolar a 4 micromolar in vitro. Eliminación: Carfilzomib tiene una vida media de ≤ 1 hora en el Día 1 del Ciclo 1 después de las dosis intravenosas ≥ 15 mg/m2. La vida media fue similar cuando se administró mediante una infusión de 30 minutos o una infusión de 2 a 10 minutos. El aclaramiento sistémico osciló entre 151 L/hora y 263 L/hora. Metabolismo: El carfilzomib se metaboliza rápidamente por la escisión de la peptidasa y la hidrólisis del epóxido fueron las principales vías del metabolismo. Los mecanismos mediados por el citocromo P450 (CYP) contribuyeron con un papel menor en el metabolismo general de carfilzomib. Excreción: Aproximadamente el 25% de la dosis administrada de carfilzomib se excreta en la orina como metabolitos en 24 horas. La excreción urinaria y fecal del compuesto original fue insignificante (0. 3% de la dosis total). Poblaciones Específicas: Edad (35 a 89 años), sexo, raza u origen étnico (80% blancos, 11% negros, 6% asiáticos, 3% hispanos) e insuficiencia renal leve a grave (depuración de creatinina de 15 mL/min a 89 mL/min) no tuvo efectos clínicamente significativos en la farmacocinética de carfilzomib. Pacientes con insuficiencia hepática: En comparación con los pacientes con función hepática normal, los pacientes con insuficiencia hepática leve (bilirrubina total 1 a 1. 5 x ULN y cualquier AST o bilirrubina total ≤ ULN y AST > ULN) e insuficiencia hepática moderada (bilirrubina total > 1. 5 a 3 x ULN y cualquier AST) tenían un ABC de carfilzomib aproximadamente 50% más alto. La farmacocinética de carfilzomib no se ha evaluado en pacientes con insuficiencia hepática grave (bilirrubina total > 3 × ULN y cualquier AST). Pacientes con insuficiencia renal: En relación con los pacientes con función renal normal, los pacientes con ESRD (pacientes con enfermedad renal en etapa terminal, ESRD por sus siglas en inglés) en hemodiálisis mostraron un ABC de carfilzomib 33% mayor. Dado que la depuración de las concentraciones de KYPROLIS en hemodiálisis no se ha estudiado, el medicamento debe administrarse después del procedimiento de hemodiálisis. Estudios de interacción con medicamentos: Estudios clínicos: Efecto de carfilzomib sobre la sensibilidad del sustrato CYP3A: La farmacocinética de midazolam (un sustrato sensible del CYP3A) no se vio afectada por la administración concomitante de carfilzomib. Estudios in vitro:Efecto de carfilzomib en las enzimas del citocromo P450 (CYP): Carfilzomib mostró una inhibición directa y dependiente del tiempo de CYP3A pero no indujo CYP1A2 y CYP3A4 in vitro. Efecto de los Transportadores sobre Carfilzomib: Carfilzomib es un sustrato de P-glicoproteína (P-gp) in vitro: Efecto de Carfilzomib sobre los Transportadores: Carfilzomib inhibe la P-gp in vitro. Sin embargo, dado que KYPROLIS se administra por vía intravenosa y se metaboliza ampliamente, es poco probable que la farmacocinética de KYPROLIS se vea afectada por los inhibidores o inductores de la P-gp. Mecanismo de Acción: Carfilzomib es una epoxicetona tetrapeptídica inhibidora del proteosoma que se une irreversiblemente a la región N-terminal de los residuos de treonina en los sitios activos del proteosoma 20S, el cual es la partícula proteolítica central del proteosoma 26S. Carfilzomib mostró actividades antiproliferativas y proapoptóticas in vitro en células de tumores sólidos y hematológicos. En animales, carfilzomib inhibió la actividad proteosomal en sangre y tejidos y retrasó el crecimiento tumoral en modelos de mieloma múltiple, de tumores hematológicos y de tumores sólidos. Farmacodinamia: La administración de carfilzomib por vía intravenosa resultó en la supresión de la actividad tipo quimotripsina (CT-L por sus siglas en inglés) del proteosoma medida en la sangre 1 hora después de la primera dosis. Dosis de carfilzomib ≥ 15 mg/m2 con o sin lenalidomida y dexametasona indujeron una inhibición ≥ 80% de la actividad CT-L del proteosoma. Asimismo, la administración de carfilzomib a 20 mg/m2 intravenosamente como agente único, resultó en la inhibición media de las subunidades del proteosoma polipéptido de baja masa molecular 2 (LMP2) y complejo de tipo multicatalítico endopeptidasa 1 (MECL1) en un rango de 26% al 32% y de 41% al 49%, respectivamente. La inhibición del proteosoma se mantuvo durante ≥ 48 horas posteriores a la primera dosis de carfilzomib por cada semana de dosificación. Estudios clínicos: En combinación con Lenalidomida y Dexametasona para el Tratamiento de Pacientes con Mieloma Múltiple en Recaída o Refractario ASPIRE ASPIRE fue un estudio aleatorizado, abierto, multicéntrico, de superioridad el cual evaluó la combinación de KYPROLIS con lenalidomida y dexametasona (KRd) versus lenalidomida y dexametasona solos (Rd) en pacientes con mieloma múltiple en recaída o refractario que habían recibido 1 a 3 líneas de terapia. (Una línea de terapia es un curso planeado de tratamiento [incluyendo inducción secuencial, trasplante, consolidación y/o mantenimiento] sin una interrupción por falta de eficacia, tal como recaída o enfermedad progresiva.) Los pacientes que presentaron lo siguiente, fueron excluidos del estudio: refractarios a bortezomib en el régimen más reciente, refractarios a lenalidomida y dexametasona en el régimen más reciente, que no respondieron a ningún régimen previo, depuración de creatinina < 50 mL/min, ALT/AST > 3. 5 x ULN y bilirrubina > 2 x ULN, insuficiencia cardiaca congestiva de Clases III a IV conforme a la New York Heart Association, o infarto al miocardio en el lapso de los últimos 4 meses. En el brazo KRd, se evaluó KYPROLIS en una dosis de arranque de 20 mg/m2, la cual fue aumentada a 27 mg/m2 en el Ciclo 1, Día 8 en adelante. Se administró KYPROLIS mediante una infusión de 10 minutos en los Días 1, 2, 8, 9, 15 y 16 de cada ciclo de 28 días del Ciclo 1 al 12. Sólo se dosificó KYPROLIS en los Días 1, 2, 15 y 16 de cada ciclo de 28 días a partir del Ciclo 13 y hasta el 18. Se administró Dexametasona 40 mg oral o intravenosamente en los Días 1, 8, 15 y 22 de cada ciclo. Se dieron oralmente 25 mg de lenalidomida en los Días 1 a 21 de cada ciclo de 28 días. El brazo de tratamiento Rd tuvo el mismo régimen para lenalidomida y dexametasona que el brazo de tratamiento KRd. Se administró KYPROLIS por un máximo de 18 ciclos a menos que se hubiera interrumpido de forma temprana por progresión de la enfermedad o toxicidad inaceptable. La administración de lenalidomida y dexametasona pudo continuar hasta progresión o toxicidad inaceptable. Para ambos brazos se requirió el uso concurrente de tromboprofilaxis y un inhibidor de bomba de protones, mientras que se requirió profilaxis antiviral para el brazo KRd. Los 792 pacientes en ASPIRE fueron aleatorizados 1:1 al brazo KRd o Rd. Las características demográficas y basales estuvieron bien balanceadas entre los dos brazos (ver Tabla 2). Sólo 53% de los pacientes tuvieron pruebas para mutaciones genéticas; se identificó una mutación genética de alto riesgo en 12% de pacientes en el brazo KRd y en 13% en el brazo Rd.
Los pacientes en el brazo KRd demostraron mejora en la supervivencia libre de progresión (PFS, por sus siglas en Inglés) comparado con aquellos en el brazo Rd (HR = 0. 69, con valor P bilateral = 0. 0001) según lo determinado usando los criterios de respuesta del Grupo Internacional de Trabajo sobre Mieloma (IMWG)/Grupo Europeo de Trasplante de Sangre y Médula (EBMT) por parte de un Comité de Revisión Independiente (IRC). La mediana de PFS fue 26. 3 meses en el brazo KRd versus 17. 6 meses en el brazo Rd (ver Tabla 3 y Figura 1). Se realizó un análisis preestablecido de la supervivencia global (OS por sus siglas en inglés) después de 246 muertes en el brazo de KRd y 267 muertes en el brazo de Rd. La mediana de seguimiento fue de aproximadamente 67 meses. Se observó una ventaja estadísticamente significativa en la OS en pacientes del brazo de KRd en comparación con los pacientes del brazo de Rd (ver Tabla 3).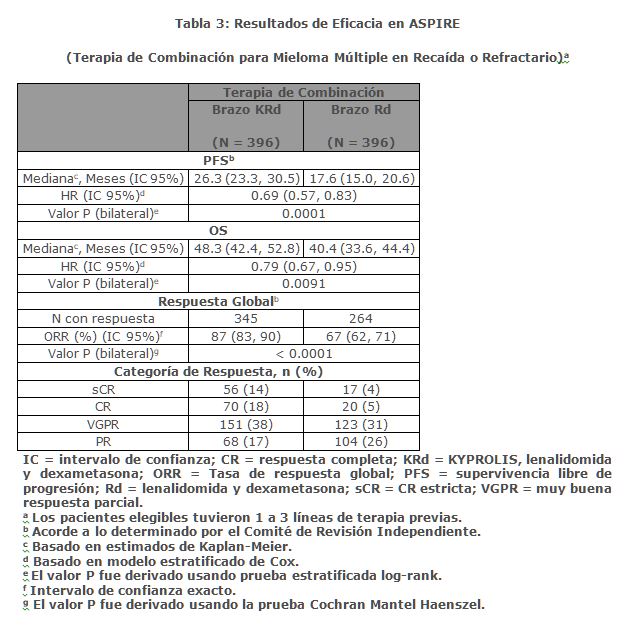
La mediana de duración de respuesta (DOR, por sus siglas en inglés) fue de 28. 6 meses (IC 95%: 24. 9, 31. 3) para los 345 pacientes que alcanzaron una respuesta en el brazo KRd y 21. 2 meses (IC 95%: 16. 7, 25. 8) para los 264 pacientes que alcanzaron una respuesta en el brazo Rd. La mediana de tiempo para respuesta fue 1 mes (rango de 1 a 14 meses) en el brazo KRd y 1 mes (rango 1 a 16 meses) en el brazo Rd.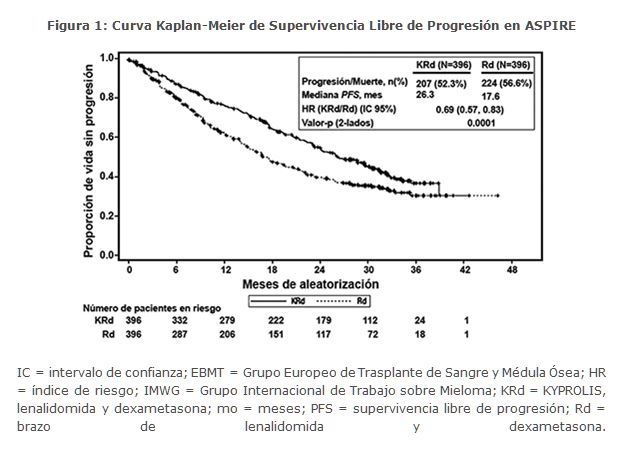
Nota: La respuesta y los resultados de PD fueron determinados usando los criterios de respuesta objetiva estándar IMWG/EBMT.
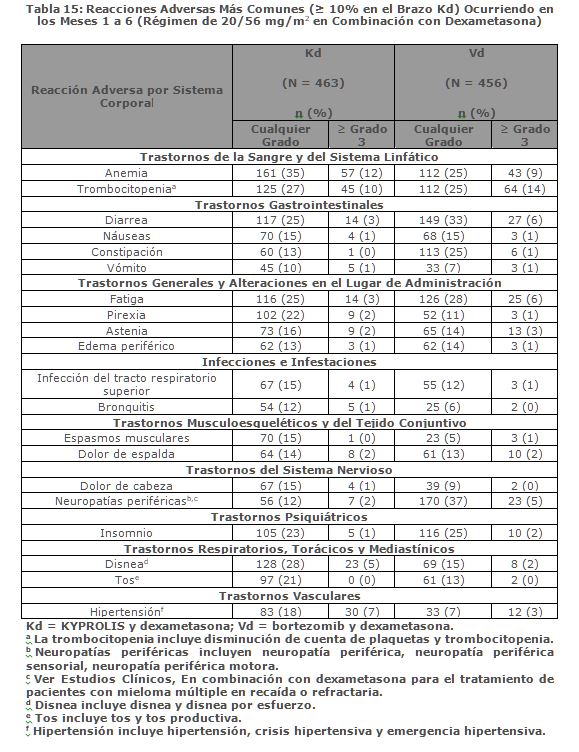
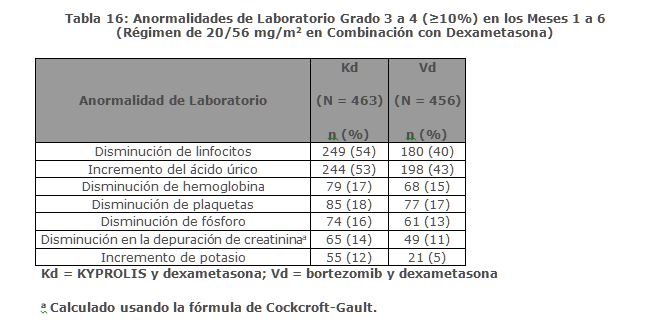
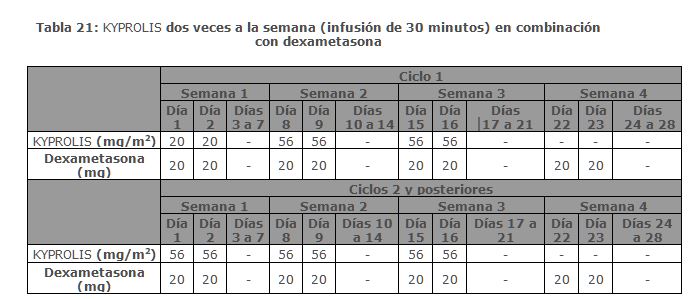
En Combinación con Dexametasona para el Tratamiento de Pacientes con Mieloma Múltiple en Recaída o Refractario: ENDEAVOR ENDEAVOR fue un estudio aleatorizado, abierto, multicéntrico, de superioridad de KYPROLIS más dexametasona (Kd) versus bortezomib más dexametasona (Vd) en pacientes con mieloma múltiple en recaída o refractario que han recibido 1 a 3 líneas de terapia. Un total de 929 pacientes fueron enrolados y aleatorizados (464 en el brazo Kd; 465 en el brazo Vd). La aleatorización fue estratificada por terapia previa de inhibidor de proteosoma (sí versus no), líneas de terapia previas (1 versus 2 o 3), etapa vigente del Sistema Internacional de Estadificación (1 versus 2 o 3) y vía planeada de administración de bortezomib. Los pacientes fueron excluidos si tuvieron menos de una respuesta parcial PR a todos los regímenes previos; depuración de creatinina < 15 mL/min; transaminasas hepáticas ≥ 3 x ULN; o fracción de eyección ventricular izquierda < 40% u otra condición cardiaca significativa. Este estudio evaluó KYPROLIS en una dosis de arranque de 20 mg/m2, la cual se incrementó a 56 mg/m2 en el Ciclo 1, del Día 8 en adelante. KYPROLIS fue administrado dos veces semanalmente mediante una infusión de 30 minutos en los Días 1, 2, 8, 9, 15 y 16 de cada ciclo de 28 días. Se administró dexametasona 20 mg oral e intravenoso en los Días, 1, 2, 8, 9, 15, 16, 22 y 23 de cada ciclo. En el brazo Vd, se dosificó bortezomib a 1. 3 mg/m2 intravenoso y subcutáneo en los Días 1, 4, 8 y 11 de un ciclo de 21 días, y se administró dexametasona 20 mg oral o intravenoso en los Días 1, 2, 4, 5, 8, 9, 11 y 12 de cada ciclo. El uso concurrente de tromboprofilaxis fue opcional, y se requirió profilaxis con un agente antiviral y un inhibidor de bomba de protones. De los 465 pacientes en el brazo Vd, 381 recibieron bortezomib subcutáneo. El tratamiento continuó hasta progresión de la enfermedad o toxicidad inaceptable. Las características demográficas y basales están resumidas en la Tabla 3.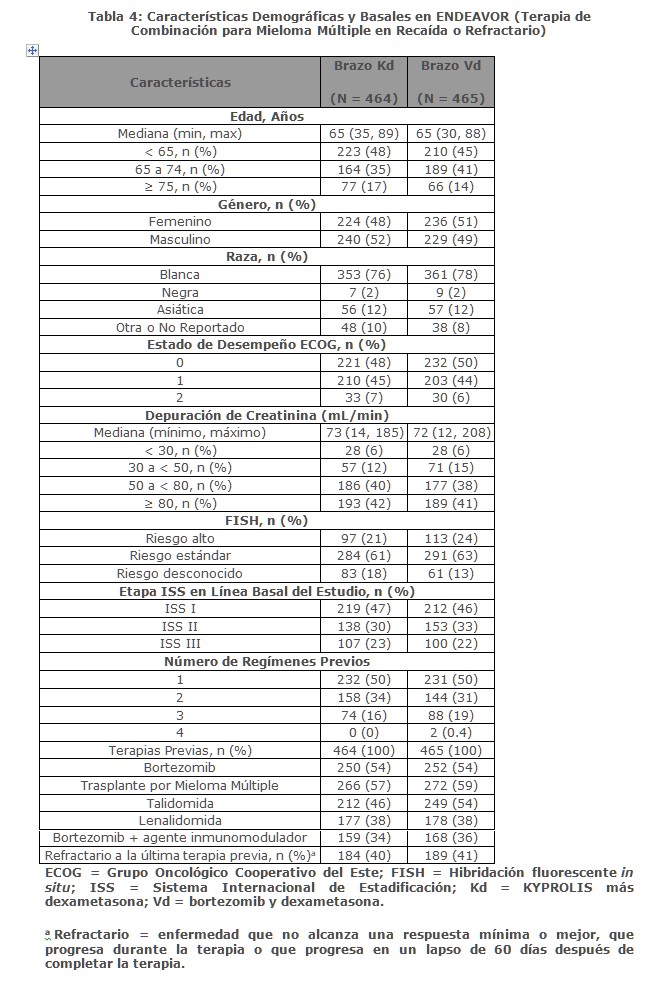
La eficacia de KYPROLIS fue evaluada mediante PFS acorde a lo determinado por un IRC usando los criterios de respuesta IMWG. El estudio mostró una mediana de PFS de 18. 7 meses en el brazo Kd versus 9. 4 meses en el brazo Vd (ver Tabla 5).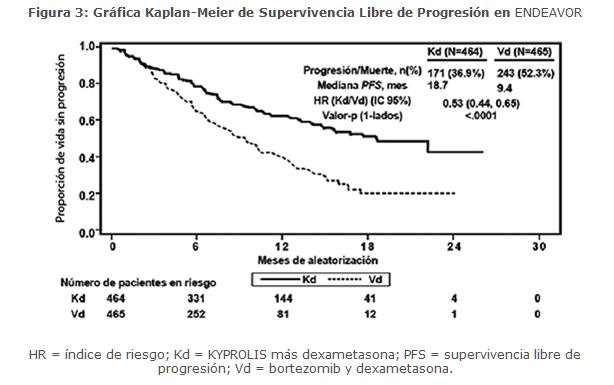
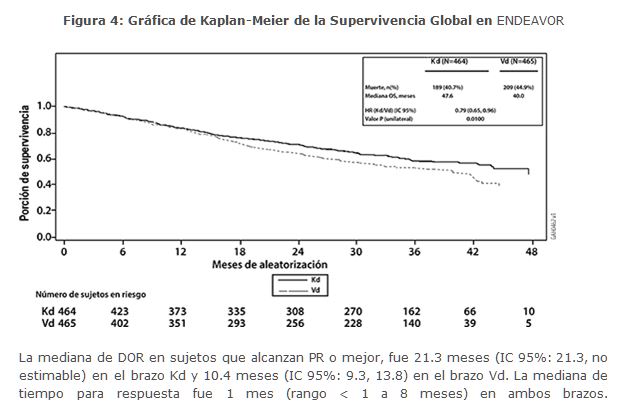
Otros criterios de valoración incluyeron la OS y la tasa de respuesta global (ORR, por sus siglas en inglés). Se realizó un análisis de la OS pre-planificado después de 189 muertes en el brazo de Kd y 209 muertes en el brazo de Vd. La mediana de seguimiento fue de aproximadamente 37 meses. Se observó una OS significativamente más prolongada en los pacientes del brazo de Kd en comparación con los pacientes en el brazo de Vd (HR = 0. 79; IC del 95%: 0. 65, 0. 96; valor de p = 0. 01) (ver la Tabla 5). El ORR fue 77% para pacientes en el brazo de Kd y 63% para pacientes en el brazo de Vd (ver Tabla 5).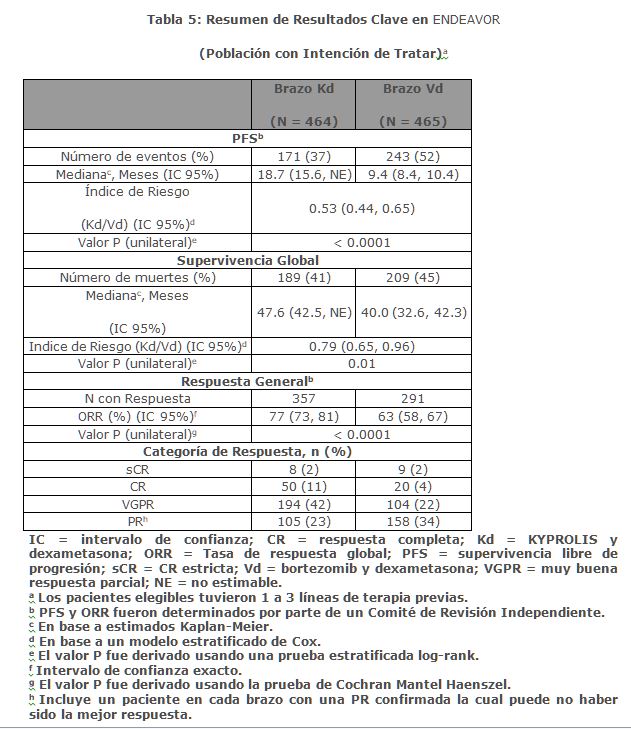
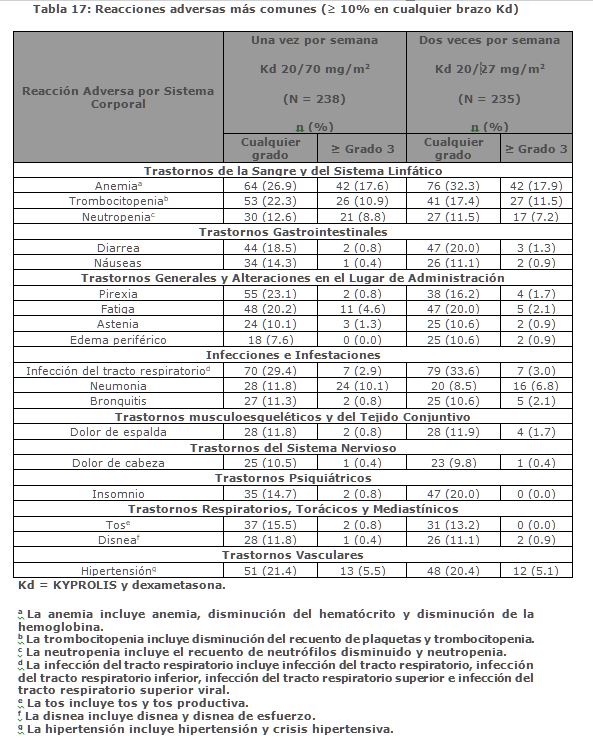
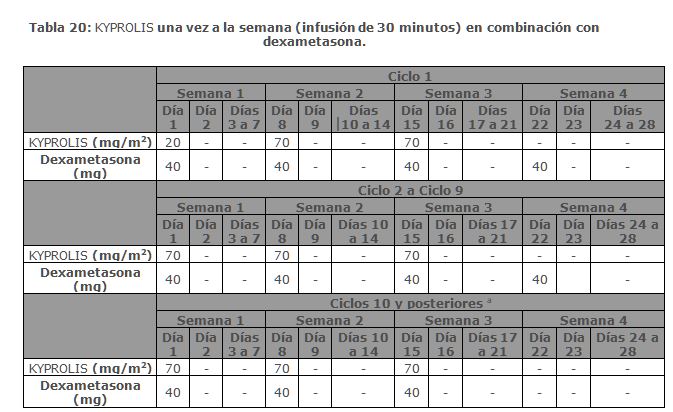
A. R. R. O. W.: A. R. R. O. W. fue un estudio aleatorizado, abierto, multicéntrico, de superioridad de KYPROLIS más dexametasona (Kd) una vez por semana (20/70 mg/m2) versus Kd dos veces por semana (20/27 mg/m2) en pacientes con mieloma múltiple en recaída y refractario que habían recibido 2 a 3 líneas previas de terapia. Los pacientes se excluyeron si tenían menos de PR (remisión parcial, por sus siglas en inglés) en al menos una línea previa; depuración de creatinina < 30 mL/min; transaminasas hepáticas ≥ 3 × ULN; o fracción de eyección del ventrículo izquierdo < 40% u otras afecciones cardiacas significativas. Un total de 478 pacientes fueron reclutados y aleatorizados (240 en el brazo de 20/70 mg/m2; 238 en el brazo de 20/27 mg/m2). La aleatorizazión se estratificó en estadios por el actual Sistema Internacional de clasificación de estadios (estadio 1 versus estadios 2 o 3), refractaria al tratamiento con bortezomib (sí versus no) y edad ( < 65 versus ≥ 65 años). El brazo 1 de este estudio evaluó KYPROLIS a una dosis inicial de 20 mg/m2, que se incrementó a 70 mg/m2 en el Ciclo 1, desde el día 8 en adelante. En el brazo 1 KYPROLIS se administró una vez a la semana mediante una infusión de 30 minutos los días 1, 8 y 15 de cada ciclo de 28 días. El brazo 2 de este estudio evaluó KYPROLIS a una dosis inicial de 20 mg/m2, que se incrementó a 27 mg/m2 en el Ciclo 1, desde el día 8 en adelante. En el brazo 2 KYPROLIS se administró dos veces por semana mediante una infusión de 10 minutos en los días 1, 2, 8, 9, 15 y 16 de cada ciclo de 28 días. En ambos regímenes, se administraron 40 mg de dexametasona por vía oral o intravenosa en los días 1, 8, 15 para todos los ciclos y en el día 22 para los ciclos 1 a 9 solamente. El uso simultáneo de tromboprofilaxis fue opcional, se recomendó la profilaxis con un agente antiviral y se requirió la profilaxis con un inhibidor de la bomba de protones. El tratamiento continuó hasta la progresión de la enfermedad o una toxicidad inaceptable. Las características demográficas y basales se resumen en la Tabla 6.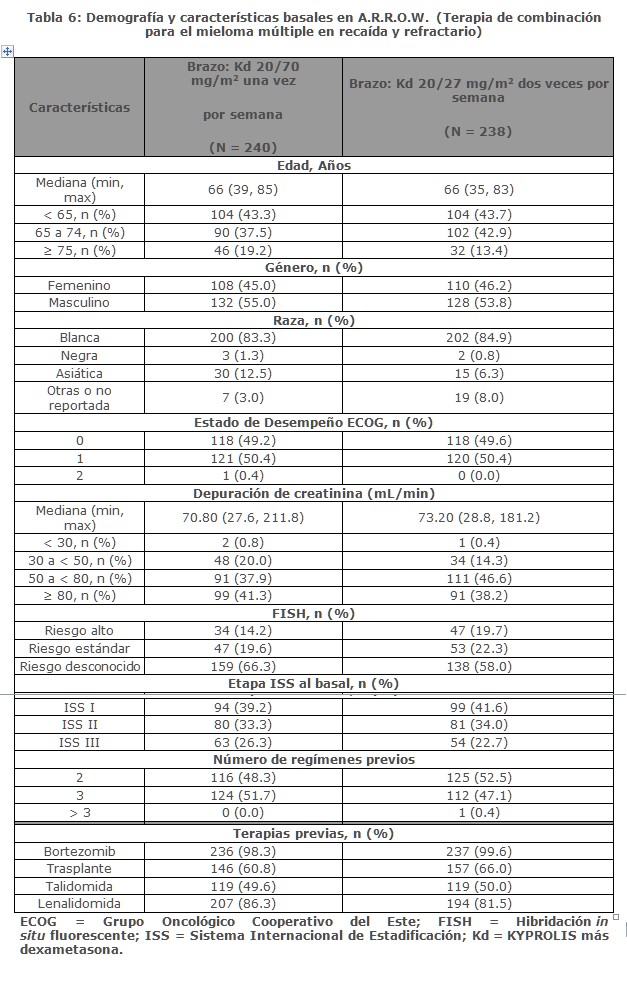
La eficacia de KYPROLIS se evaluó por la PFS utilizando los criterios de respuesta del IMWG. El estudio mostró una mediana de PFS de 11. 2 meses en el brazo de Kd 20/70 mg/m2 una vez a la semana frente a 7. 6 meses en el brazo de Kd 20/27 mg/m2 dos veces por semana (consulte la Tabla 7).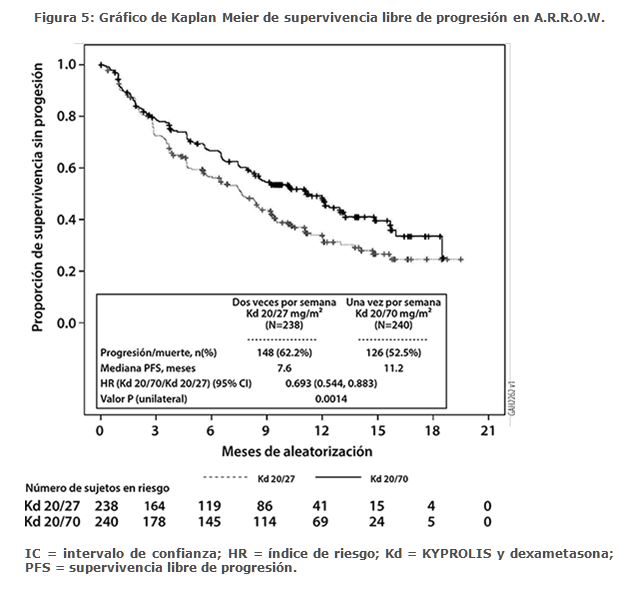
Entre otros objetivos del estudio se incluyó la ORR. La ORR fue de 62. 9% para los pacientes en el brazo de Kd 20/70 mg/m2 una vez por semana y del 40. 8% para los pacientes en el brazo de Kd 20/27 mg/m2 dos veces por semana (consulte la Tabla 7).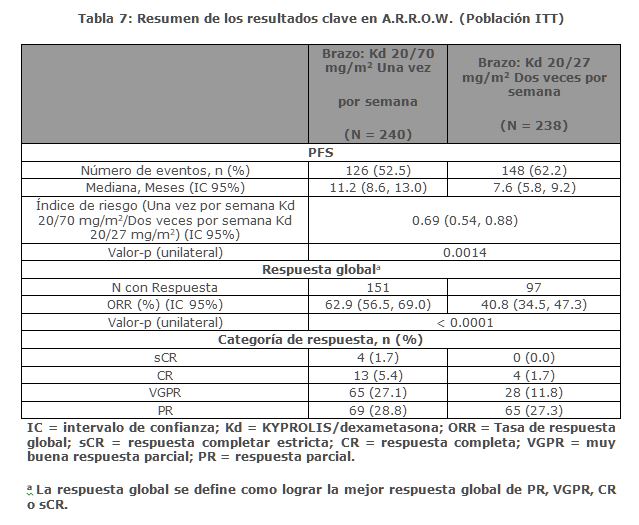
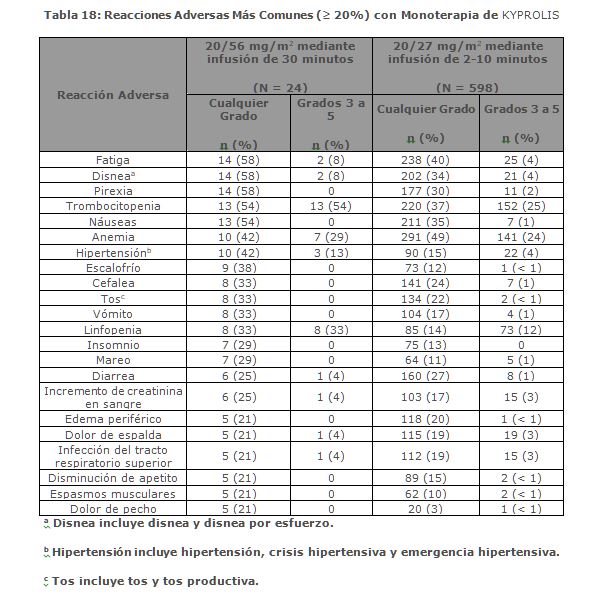
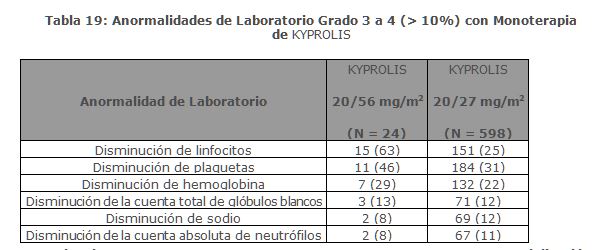
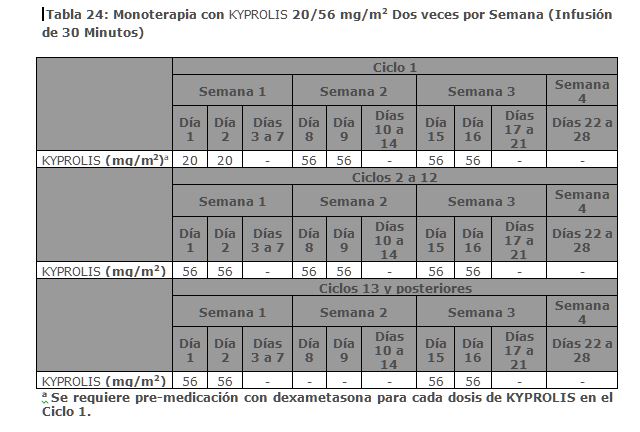
La mediana de DOR en sujetos que alcanzaron PR o mejor fue de 15 meses (IC 95%: 12. 2, no estimable) en el brazo de Kd 20/70 mg/m2 y 13. 8 meses (IC 95%: 9. 5, no estimable) en el brazo de Kd 20/27 mg/m2. El tiempo medio de respuesta fue de 1. 1 meses en el brazo de Kd 20/70 mg/m2 y de 1. 9 meses en el brazo de Kd 20/27 mg/m2. Monoterapia para el Tratamiento de Pacientes con Mieloma Múltiple en Recaída o Refractario: Estudio PX-171-007: El estudio PX-171-007 fue un estudio multicéntrico, abierto, de escalamiento de dosis, de brazo único que evaluó la seguridad de la monoterapia con carfilzomib mediante una infusión de 30 minutos en pacientes con mieloma múltiple en recaída o refractario después de 2 o más líneas de terapia. Los pacientes fueron excluidos si tenían una depuración de creatinina < 20 mL/min; ALT ≥ 3 x límite superior del normal (ULN); bilirrubina ≥ 1. 5 x ULN; insuficiencia cardiaca congestiva de clases III o IV de la New York Heart Association, u otra condición cardiaca significativa. Un total de 24 sujetos con mieloma múltiple fueron enrolados en el nivel de dosis máxima tolerada de 20/56 mg/m2. Se administró carfilzomib dos veces semanalmente por 3 semanas consecutivas (Días 1, 2, 8, 9, 15 y 16) de un ciclo de 28 días. Del Ciclo 13 en adelante, las dosis de carfilzomib de los días 8 y 9 pudieron ser omitidas. Los pacientes recibieron carfilzomib en una dosis de arranque de 20 mg/m2 en los Días 1 y 2 del Ciclo 1, la cual se incrementó a 56 mg/m2 para todas las dosis subsecuentes. Se requirió dexametasona 8 mg oral o intravenoso antes de cada dosis de carfilzomib en el Ciclo 1 y fue opcional en ciclos subsecuentes. Se continuó el tratamiento hasta progresión de la enfermedad o toxicidad inaceptable. La eficacia fue evaluada mediante ORR y DOR. La ORR por evaluación del investigador fue 50% (IC 95%: 29, 71) acorde a los criterios IMWG (ver Tabla 8). La mediana de DOR en sujetos que alcanzaron una PR o mejor fue 8. 0 meses (Rango: 1. 4, 32. 5).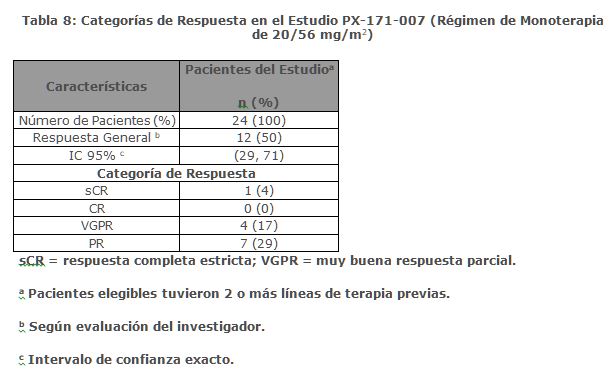
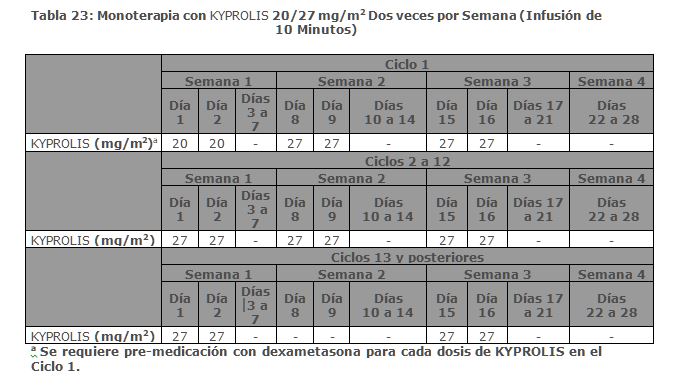
Estudio PX-171-003 A1: El Estudio PX-171-003 A1, fue un estudio de un solo brazo, multicéntrico de monoterapia con KYPROLIS mediante infusión de hasta 10 minutos. Los pacientes elegibles fueron aquellos con mieloma múltiple en recaída y refractario que habían sido sometidos al menos a dos tratamientos previos (incluido el tratamiento con bortezomib y talidomida y/o lenalidomida) y tuvieron ≤ 25% de respuesta a la terapia más reciente o tuvieron progresión de la enfermedad durante o en un lapso de 60 días de la terapia más reciente. Los pacientes excluidos del ensayo fueron aquellos refractarios a las terapias anteriores o con bilirrubina total ≥ 2 x ULN; depuración de creatinina < 30 mL/min; insuficiencia cardiaca congestiva Clases III a IV conforme a la New York Heart Association (NYHA, por sus siglas en Inglés); isquemia cardiaca sintomática; infarto de miocardio dentro de los últimos 6 meses; neuropatía periférica Grado 3 o 4; o neuropatía periférica Grado 2 con dolor; infecciones activas que requerían tratamiento; o derrame pleural. Se administró KYPROLIS por vía intravenosa hasta 10 minutos en dos días consecutivos a la semana por tres semanas, seguido de un periodo de descanso de 12 días (ciclo de tratamiento de 28 días), hasta la progresión de la enfermedad, toxicidad inaceptable o por un máximo de 12 ciclos. Los pacientes recibieron 20 mg/m2 por dosis en el Ciclo 1, y 27 mg/m2 en los ciclos subsecuentes. Se administró dexametasona de 4 mg por vía oral o intravenosa antes de la administración de las dosis de KYPROLIS en el primer y segundo ciclo. Un total de 266 pacientes fueron enrolados. Las características basales de los pacientes y de la enfermedad se resumen en la Tabla 9.
La eficacia fue evaluada mediante ORR acorde a lo determinado por la evaluación del IRC usando los criterios del IMWG. La mediana del número de ciclos iniciados fue cuatro. La ORR (PR o mejor) fue del 23% (IC 95%: 18, 28) (ver Tabla 10). La mediana de la DOR fue de 7. 8 meses (IC 95%: 5. 6, 9. 2).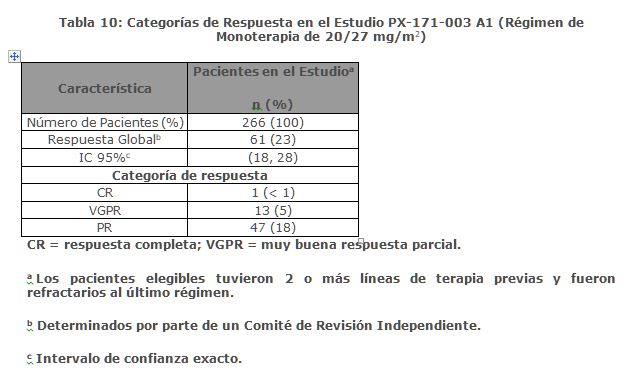
Estudio PX-171-004 Parte 2: El estudio PX-171-004 Parte 2 fue un estudio clínico de un solo brazo, multicéntrico de monoterapia con KYPROLIS mediante infusión de hasta 10 minutos. Los pacientes elegibles fueron aquellos con mieloma múltiple en recaída o refractario que no tuvieron tratamientos previos con bortezomib, habían recibido de una a tres líneas de terapia previas y tuvieron ≤ 25% de respuesta o progresión durante la terapia o progresión en el lapso de 60 días después del término de la terapia. Los pacientes fueron excluidos del estudio si eran refractarios a la terapia estándar de primera línea o tenían bilirrubina total ≥ 2 × ULN; depuración de creatinina < 30 mL/min; falla cardiaca congestiva Clases III o IV conforme a la New York Heart Association; isquemia cardiaca sintomática; infarto al miocardio dentro de los últimos 6 meses; infecciones activas que requieren tratamiento; o derrame pleural. Se administró KYPROLIS intravenosamente hasta 10 minutos en dos días consecutivos cada semana por tres semanas, seguido por un periodo de descanso de 12 días (ciclo de tratamiento de 28 días), hasta la progresión de la enfermedad, toxicidad inaceptable, o por un máximo de 12 ciclos. Los pacientes recibieron 20 mg/m2 en cada dosis en el Ciclo 1, y 27 mg/m2 en ciclos subsecuentes. Se administró dexametasona 4 mg oral o intravenosa antes de las dosis de KYPROLIS en el primero y segundo ciclo. Se trataron un total de 70 pacientes con este régimen de 20/27 mg/m2. Las características basales del paciente y de la enfermedad se resumen en la Tabla 11.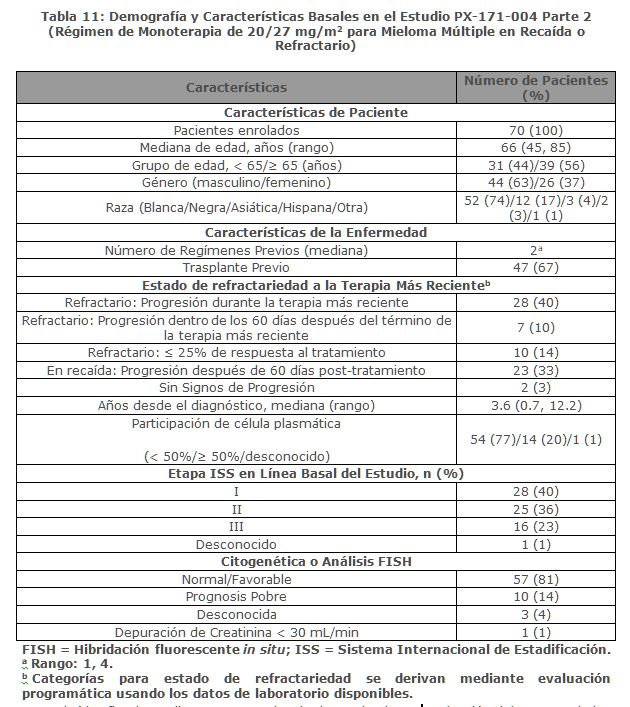
Se evaluó la eficacia mediante ORR acorde a lo determinado por evaluación del IRC usando los criterios del IMWG. La mediana del número de ciclos iniciados fue siete. La ORR (PR o mejor) fue 50% (IC 95%: 38, 62) (ver Tabla 12). No se alcanzó la mediana de la DOR.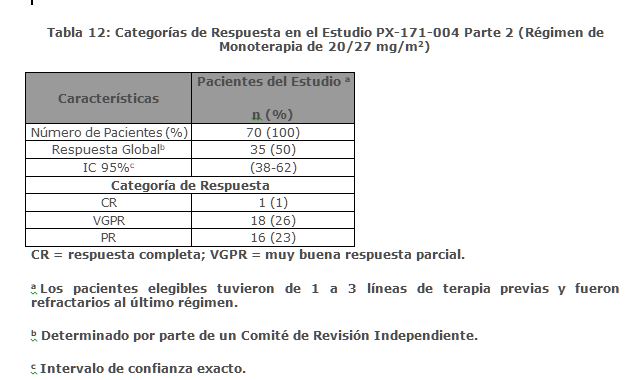
Contraindicaciones: KYPROLIS está contraindicado en pacientes con hipersensibilidad a carfilzomib o sus derivados, o cualquier excipiente en la formulación.
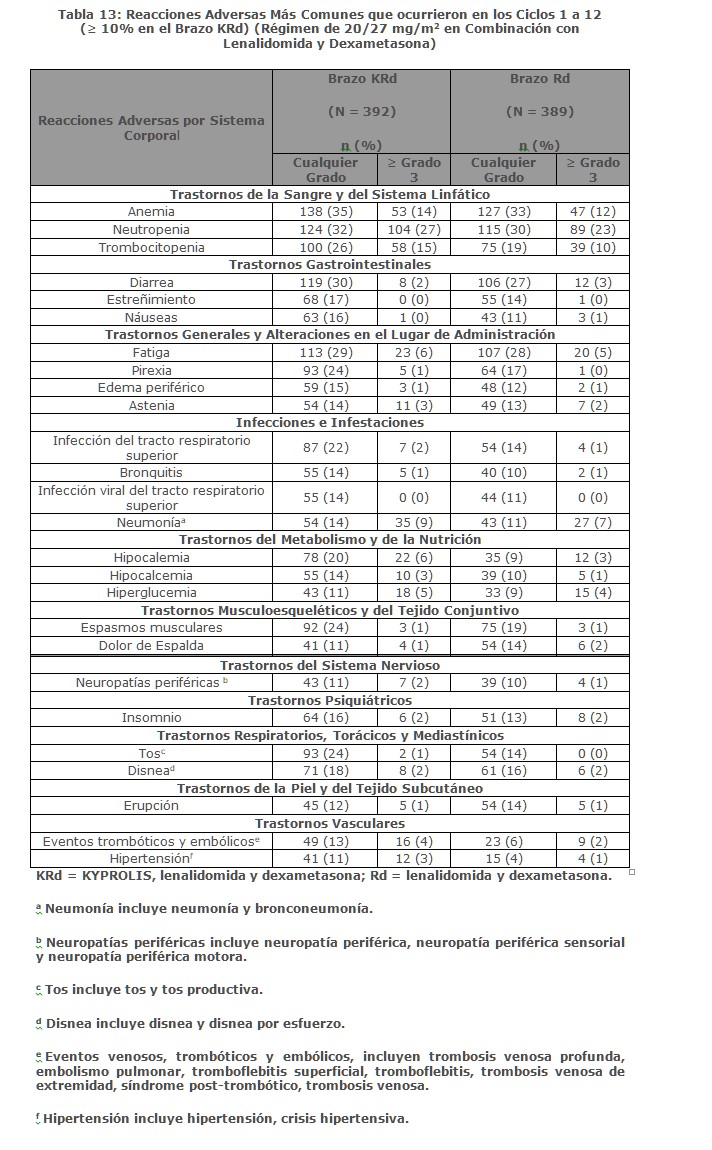
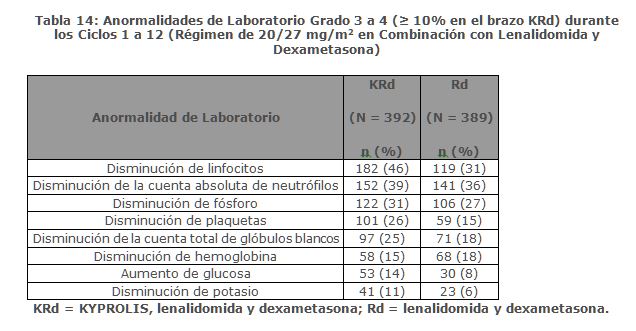
Precauciones generales: Toxicidades Cardiacas: Se han producido nuevos casos o empeoramiento de insuficiencia cardiaca pre-existente (por ejemplo, insuficiencia cardiaca congestiva, edema pulmonar, disminución en la fracción de eyección), cardiomiopatía restrictiva, isquemia e infarto al miocardio incluyendo fatalidades, posteriores a la administración de KYPROLIS. Algunos eventos ocurrieron en pacientes con función ventricular basal normal. En estudios clínicos con KYPROLIS, estos eventos ocurrieron durante el curso de la terapia con KYPROLIS. Ha ocurrido muerte por paro cardiaco a un día de la administración de KYPROLIS. En estudios aleatorizados, abiertos, multicéntricos para terapias de combinación, la incidencia de eventos por falla cardiaca fue 8% (ver Reacciones secundarias y adversas). Monitorizar a los pacientes para detectar signos o síntomas clínicos de insuficiencia cardiaca o isquemia cardiaca. Evaluar inmediatamente si se sospecha de toxicidad cardiaca. Interrumpir el tratamiento con KYPROLIS en caso de eventos adversos cardiacos Grado 3 o 4, hasta que se alcance la recuperación y evaluar si reiniciar el tratamiento con KYPROLIS con una reducción de dosis con base en una evaluación de beneficio/riesgo (ver Dosis y vía de administración). Mientras que por un lado se requiere hidratación adecuada previa a cada dosificación en el Ciclo 1, por otro lado, todos los pacientes también deberán ser monitoreados para detectar evidencia de sobrecarga de volumen, especialmente pacientes en riesgo de insuficiencia cardiaca. Ajustar la ingesta total de fluidos según sea lo apropiado clínicamente, en pacientes con insuficiencia cardiaca basal o quienes estén en riesgo de insuficiencia cardiaca (ver Dosis y vía de administración). El riesgo de falla cardiaca se incrementa en pacientes ≥ 75 años de edad en comparación con los pacientes < 75 años de edad. El riesgo de falla cardiaca también aumenta en pacientes asiáticos. En los ensayos clínicos no participaron pacientes con insuficiencia cardiaca Clase III y IV conforme a la NYHA, infarto al miocardio reciente, y aquellos con alteraciones en la conducción, angina o arritmias no controladas con fármacos. Estos pacientes pueden estar en mayor riesgo de complicaciones cardiacas por lo que deben ser sometidos a evaluación médica integral (incluyendo el control de la presión sanguínea y el manejo de líquidos) antes de empezar el tratamiento con KYPROLIS o bien permanecer bajo un seguimiento cercano [ver Uso en poblaciones específicas (Uso geriátrico)]. Falla Renal Aguda: Se han producido casos de falla renal aguda en pacientes que recibieron KYPROLIS. Algunos de estos eventos han sido fatales. Los eventos adversos de insuficiencia renal (incluyendo falla renal) han ocurrido en aproximadamente 11% de los pacientes tratados con KYPROLIS. La falla renal aguda fue reportada con mayor frecuencia en pacientes con mieloma múltiple avanzado en recaída y refractario que recibieron monoterapia con KYPROLIS. El riesgo de falla renal fatal fue mayor en pacientes con una depuración de creatinina estimada basal reducida (calculada utilizado la ecuación de Cockcroft y Gault). Monitorizar la función renal con mediciones regulares de creatinina sérica y/o estimado de depuración de creatinina. Reducir o interrumpir la dosis según sea apropiado (ver Dosis y vía de administración). Síndrome de Lisis Tumoral: Se han reportado casos de síndrome de lisis tumoral (TLS por sus siglas en inglés), incluyendo resultados fatales, en pacientes que recibieron KYPROLIS. Pacientes con mieloma múltiple y elevada carga tumoral deben ser considerados con mayor riesgo de TLS. Asegurarse que los pacientes se encuentren bien hidratados antes de la administración de KYPROLIS en el Ciclo 1 y en los ciclos subsecuentes según se requiera (ver Dosis y vía de administración). Considerar el uso de fármacos para reducir el ácido úrico en pacientes con alto riesgo para TLS. Monitorizar manifestaciones de TLS durante el tratamiento y de presentarse resolverlo de inmediato incluyendo la interrupción del tratamiento con KYPROLIS hasta la resolución del TLS (ver Dosis y vía de administración). Toxicidad Pulmonar:Han ocurrido casos de Síndrome de Dificultad Respiratoria Aguda (ARDS, por sus siglas en Inglés), insuficiencia respiratoria aguda, y enfermedad pulmonar infiltrativa difusa aguda tal como neumonitis y enfermedad pulmonar intersticial en aproximadamente el 1% de los pacientes que reciben KYPROLIS. Algunos eventos han sido fatales. En la eventualidad de toxicidad pulmonar inducida por fármaco, discontinuar KYPROLIS (ver Dosis y vía de administración). Hipertensión Pulmonar:Se reportó hipertensión arterial pulmonar en aproximadamente 1% de los pacientes tratados con KYPROLIS y fue Grado 3 o mayor en menos del 1% de los pacientes. Evaluar con escaneo cardiaco y/u otras pruebas según lo indicado. Interrumpir el uso de KYPROLIS en caso de hipertensión pulmonar hasta que esta condición se haya resuelto o haya regresado a condiciones basales y considerar si se reinicia KYPROLIS con base a una valoración de beneficio/riesgo (ver Dosis y vía de administración). Disnea: La disnea fue reportada en el 28% de los pacientes tratados con KYPROLIS y fue Grado 3 o mayor en el 4% de los pacientes. Evaluar los casos de disnea para excluir condiciones cardiopulmonares incluyendo falla cardiaca y síndromes pulmonares. Interrumpir el uso de KYPROLIS en los casos de disnea Grado 3 y 4 hasta que se resuelvan o hayan regresado a condiciones basales. Considerar si se reinicia KYPROLIS con base a una valoración del riesgo/beneficio [ver Dosis y vía de administración y Reacciones secundarias y adversas (Experiencia en estudios clínicos)]. Hipertensión:La hipertensión, incluyendo crisis y emergencia hipertensiva, ha sido observada con KYPROLIS. En un estudio aleatorizado, abierto, multicéntrico, que evaluó KRd versus Rd, la incidencia de eventos de hipertensión fue del 17% en el brazo KRd versus 9% en el brazo Rd. En un estudio aleatorizado, abierto, multicéntrico de Kd versus Vd, la incidencia de eventos de hipertensión fue 34% en el brazo Kd versus 11% en el brazo Vd. Algunos de estos eventos han sido fatales. Se recomienda el control de hipertensión antes de iniciar con KYPROLIS. Monitorizar la presión sanguínea regularmente en todos los pacientes en tratamiento con KYPROLIS. Si la hipertensión no puede ser controlada adecuadamente, se deberá interrumpir KYPROLIS y evaluar. Considerar si se reinicia KYPROLIS con base en evaluación de beneficio/riesgo (ver Dosis y vía de administración). Trombosis Venosa: Eventos tromboembólicos venosos (incluyendo trombosis venosa profunda y embolismo pulmonar) han sido observados con KYPROLIS. En un estudio aleatorizado, abierto, multicéntrico, evaluando KRd versus Rd (con tromboprofilaxis usada en ambos brazos), la incidencia de eventos tromboembólicos venosos en los primeros 12 ciclos fue de 13% en el brazo KRd versus 6% en el brazo Rd. En un estudio aleatorizado, abierto, multicéntrico de Kd versus Vd, la incidencia de eventos tromboembólicos venosos en los meses 1 a 6 fue de 9% en el brazo Kd versus 2% en el brazo Vd. Con la monoterapia de KYPROLIS, la incidencia de eventos tromboembólicos venosos fue 2%. La tromboprofilaxis es recomendada para pacientes que están siendo tratados con la combinación de KYPROLIS con dexametasona o con lenalidomida más dexametasona. El régimen de tromboprofilaxis debe estar basado en la evaluación de los riesgos subyacentes de los pacientes. Los pacientes que usan anticonceptivos orales o un método hormonal de anticoncepción asociado con un riesgo de trombosis, deberán considerar un método alterno y efectivo de anticoncepción durante el tratamiento con KYPROLIS en combinación con dexametasona o lenalidomida más dexametasona (ver Restricciones de uso durante el embarazo y la lactancia. Reacciones a la Infusión:Han ocurrido reacciones a la infusión incluyendo reacciones potencialmente mortales en pacientes recibiendo KYPROLIS. Los síntomas incluyen fiebre, escalofríos, artralgia, mialgia, enrojecimiento facial, edema facial, vómito, debilidad, dificultad para respirar, hipotensión, síncope, dolor torácico o angina. Estas reacciones pueden ocurrir inmediatamente después o dentro de las 24 horas posteriores a la administración de KYPROLIS. Administrar dexametasona previo a la administración de KYPROLIS para reducir la incidencia e intensidad de las reacciones a la infusión (ver Dosis y vía de administración). Informar a los pacientes del riesgo y de los síntomas y que debe contactar a su médico inmediatamente si ocurren síntomas de reacción a la infusión. Hemorragia: Se han reportado casos de hemorragia grave o fatal en pacientes tratados con KYPROLIS. Los eventos hemorrágicos han incluido hemorragia gastrointestinal, pulmonar e intracraneal y epistaxis. El sangrado puede ser espontáneo, y la hemorragia intracraneal ha ocurrido sin trauma. Se ha reportado hemorragia en pacientes con recuentos plaquetarios bajos o normales. La hemorragia también se ha reportado en pacientes que no estaban en terapia antiplaquetaria o anticoagulación. Evaluar inmediatamente los signos y síntomas de pérdida de sangre. Reducir o suspender la dosis según sea apropiado (ver Dosis y vía de administración y Reacciones secundarias y adversas). Trombocitopenia: KYPROLIS causa trombocitopenia con nadir plaquetario que se observa entre el día 8 y el día 15 de cada ciclo de 28 días, con una recuperación en la cuenta de plaquetas a cifras basales que ocurre usualmente al inicio del ciclo subsecuente (ver Reacciones secundarias y adversas). Se reportó trombocitopenia en aproximadamente 32% de los pacientes en los estudios clínicos con KYPROLIS. Monitorear frecuentemente el recuento de plaquetas durante el tratamiento con KYPROLIS. Reducir o suspender la dosis según sea apropiado (ver Dosis y vía de administración). Puede ocurrir hemorragia (ver Reacciones secundarias y adversas). Toxicidad Hepática y Falla Hepática:Se han reportado casos de insuficiencia hepática, incluyendo casos mortales ( < 1%) durante el tratamiento con KYPROLIS. KYPROLIS puede causar incremento en los niveles de transaminasas séricas. Monitorizar regularmente las enzimas hepáticas independientemente de los valores basales. Reducir o interrumpir la dosis según sea apropiado (ver Dosis y vía de administración y Reacciones secundarias y adversas). Microangiopatía Trombótica:Se han reportado casos de microangiopatía trombótica, incluyendo púrpura trombocitopénica trombótica/síndrome hemolítico urémico (TTP/HUS, por sus siglas en Inglés) en pacientes que han recibido KYPROLIS. Algunos de estos eventos han sido fatales. Monitorizar signos y síntomas de TTP/HUS. Si se sospechan estos diagnósticos, se deberá suspender KYPROLIS y evaluar. Si se excluye el diagnóstico de TTP/HUS, se puede reiniciar el tratamiento con KYPROLIS. Se desconoce la seguridad en pacientes con TTP/HUS al reiniciar la terapia con KYPROLIS (ver Dosis y vía de administración). Síndrome de Encefalopatía Posterior Reversible: Se han reportado casos de síndrome de encefalopatía posterior reversible (PRES, por sus siglas en Inglés) en pacientes recibiendo KYPROLIS. PRES, previamente denominado Síndrome de Leucoencefalopatía Posterior Reversible (RPLS), es un trastorno neurológico, el cual puede presentarse con convulsiones, cefalea, letargia, confusión, ceguera, alteración de la conciencia, y otras alteraciones visuales y neurológicas, además de hipertensión, y el diagnóstico es confirmado por medio de imágenes neuro-radiológicas (MRI). Interrumpir KYPROLIS si se sospecha de PRES y evaluar. Se desconoce la seguridad al reiniciar la terapia con KYPROLIS en pacientes que previamente experimentaron PRES. Incremento en las Toxicidades Fatales y Graves en Combinación con Melfalán y Prednisona en Pacientes Recientemente Diagnosticados No elegibles para Trasplante: En un estudio clínico de 955 pacientes no elegibles para trasplante con mieloma múltiple de reciente diagnóstico aleatorizados a KYPROLIS (20/36 mg/m2 con infusión de 30 minutos dos veces a la semana por cuatro de cada ciclo de seis semanas), melfalán y prednisona (KMP) o bortezomib, melfalán y prednisona (VMP), se observó una mayor incidencia de reacciones adversas fatales (7% versus 4%) y reacciones adversas graves (50% versus 42%) en el brazo KMP comparado con los pacientes en el brazo VMP, respectivamente. Se observó que los pacientes en el brazo KMP tuvieron una mayor incidencia de reacciones adversas de cualquier grado incluyendo falla cardiaca (11% versus 4%), hipertensión (25% versus 8%), falla renal aguda (14% versus 6%) y disnea (18% versus 9%). Este estudio no alcanzó su medición primaria de superioridad en la supervivencia libre de progresión (PFS) para el brazo KMP. KYPROLIS, en combinación con melfalán y prednisona no está indicado para pacientes no elegibles para trasplante con mieloma múltiple de reciente diagnóstico. Toxicidad Embrio-Fetal:Según el mecanismo de acción y los hallazgos en animales, KYPROLIS puede causar daño fetal cuando se administra a una mujer embarazada. Las mujeres con potencial reproductivo deben evitar embarazarse mientras reciben tratamiento con KYPROLIS. Si se usa KYPROLIS durante el embarazo o si la paciente se embaraza durante el tratamiento con KYPROLIS, se debe informar al paciente sobre el riesgo potencial para el feto (ver Restricciones de uso durante el embarazo y la lactancia y Precauciones en relación con efectos de carcinogénesis, mutagénesis y teratogénesis, y sobre la fertilidad). Efectos en la Habilidad para Manejar u Operar MaquinariaNo se han llevado a cabo estudios sobre los efectos de carfilzomib sobre la capacidad para conducir o utilizar maquinaria. Fatiga, mareo, desmayos y/o reducción de la presión arterial han sido observados en estudios clínicos. Por lo tanto, pacientes tratados con KYPROLIS deben de ser advertidos de no manejar ni operar maquinaria si presentan alguno de estos síntomas.
Restricciones de uso durante el embarazo y la lactancia: Embarazo:Resumen del Riesgo: KYPROLIS puede causar daño fetal en base a los hallazgos en estudios con animales (ver Precauciones en relación con efectos de carcinogénesis, mutagénesis y teratogénesis, y sobre la fertilidad) y al mecanismo de acción del medicamento (ver Mecanismo de acción). No hay estudios con el uso de KYPROLIS en mujeres embarazadas para informar los riesgos asociados con los medicamentos de desarrollar resultados adversos. KYPROLIS causó letalidad fetal embrionaria en conejos a dosis más bajas que la dosis clínica. Aconseje a las mujeres embarazadas sobre el riesgo potencial para el feto. Se desconoce el riesgo estimado de fondo de defectos mayores al nacimiento y de abortos espontáneos para la población indicada. Todos los embarazos tienen un riesgo de fondo de defectos de nacimiento, pérdida y otros resultados adversos. En la población general de EUA, el riesgo estimado de fondo de defectos congénitos importantes y abortos espontáneos clínicamente reconocidos es de 2% a 4% y de 15% a 20%, respectivamente. Lactancia: Resumen de Riesgo: No hay información en relación con la presencia de KYPROLIS en la leche materna humana, los efectos sobre el niño en lactancia, o los efectos sobre la producción de leche. Debido a que muchos medicamentos se excretan en la leche materna y se desconoce la posibilidad de reacciones adversas graves de KYPROLIS en un niño amamantado, se aconseja a las mujeres lactantes que no amamanten durante el tratamiento con KYPROLIS y durante las 2 semanas posteriores al tratamiento. Mujeres y hombres con potencial reproductivo: Basándose en su mecanismo de acción y los hallazgos en animales, KYPROLIS puede causar daño fetal cuando se administra a una mujer embarazada. Pruebas de embarazo: Realice pruebas de embarazo en mujeres con potencial reproductivo antes de iniciar el tratamiento con KYPROLIS. Anticoncepción: Mujeres: Informar a las mujeres con potencial reproductivo evitar el embarazo y que deben usar un método anticoncepti