MASENNUS
PSICOFARMA
Denominación genérica: Atomoxetina
Forma farmacéutica y formulación: Cápsulas. Cada cápsula contiene: Clorhidrato de atomoxetina equivalente a 25 mg, 40 mg y 60 mg de atomoxetina. Excipiente, cbp 1 Cápsula
Indicaciones terapéuticas: MASENNUS contiene atomoxetina, un fármaco inhibidor selectivo de la recaptura de noradrenalina, indicado en el tratamiento del trastorno por déficit de atención con hiperactividad (TDAH) en niños mayores de 6 años de edad, adolescentes y adultos.
Farmacocinética y farmacodinamia: Farmacodinamia: Atomoxetina actúa mediante la inhibición selectiva del transportador presináptico de la noradrenalina (NE). El isómero R (-) de la atomoxetina, inhibe selectivamente la recaptura neuronal de la noradrenalina (NE). Los sitios de unión de la atomoxetina en el cerebro son consistentes con la distribución conocida de las neuronas que contienen noradrenalina. La potenciación de la actividad neurotransmisora, en el sistema nervioso central (SNC) es la que condiciona la actividad antidepresiva. El aumento de las concentraciones de noradrenalina, dan lugar a la desensibilización de los receptores beta-adrenérgicos. La atomoxetina tiene mínima o ninguna afinidad por otros sitios transportadores o receptores de neurotransmisores (dopamina y serotonina). El aumento de la noradrenalina inducido por atomoxetina en la corteza prefrontal, mejora la atención y la memoria. Sobre el segmento QT y QTc, en adultos sanos con metabolizadores lentos del CYP2D6, se administró dosis de 60 mg de atomoxetina dividido en dos tomas al día, mostrando que las concentraciones máximas, no presentaron diferencia que con el placebo. Se observó un ligero incremento en el segmento QTc con el aumento de la concentración de atomoxetina. Farmacocinética: La atomoxetina presenta casi la misma farmacocinética en niños, adolescentes y adultos, pero aún no se evalúa en niños menores de 6 años. Absorción: Después de su administración oral, atomoxetina se absorbe rápido y casi completamente, alcanzando concentraciones plasmáticas máximas promedio (Cmáx) en aproximadamente 1 a 2 horas. La atomoxetina puede ser administrada con o sin la comida. Distribución: Su volumen de distribución es aproximadamente 0.85 L / kg lo que indica que la atomoxetina se distribuye principalmente en el agua corporal total. La atomoxetina se une 98% a proteínas plasmáticas, principalmente a la albúmina. Metabolismo: La atomoxetina es metabolizada principalmente a través de oxidación por la isoenzima del citocromo P-450 (CYP2D6) y posterior glucuronidación. El principal metabolito oxidativo de la atomoxetina es la 4-hidroxiatomoxetina, que es tan potente como la atomoxetina, pero circula en concentraciones mucho más bajas. Eliminación: La vida media de eliminación de atomoxetina en metabolizadores rápidos es de 3.6 horas y para metabolizadores lentos es de 21 horas tras su administración oral. La acumulación de atomoxetina ocurre en los metabolizadores lentos pero no en los metabolizadores rápidos. La biotransformación es muy amplia, por lo que < 3% de la dosis es excretada como fármaco inalterado. La atomoxetina se excreta principalmente en la orina ( > 80%) como 4 hidroxiatomoxetina-O-glucurónido y en las heces ( < 17%).
Contraindicaciones: Está contraindicado en casos de hipersensibilidad a la atomoxetina y/o a cualquier otro componente de la formulación. También está contraindicada en pacientes que se encuentren bajo tratamiento con Inhibidores de la Monoaminoxidasa (IMAO) o dentro del periodo de 2 semanas de haberlo terminado. Así como en paciente con glaucoma de ángulo cerrado. En trastornos cardiovasculares graves pueden incluir hipertensión grave, insuficiencia cardíaca, enfermedad arterial oclusiva, angina, enfermedad cardíaca congénita hemodinámicamente significativa, cardiomiopatía, infarto de miocardio, arritmias potencialmente mortales y canalopatías o en trastornos cerebrovasculares graves pueden incluir aneurisma cerebral o ictus, está contraindicada la atomoxetina. Atomoxetina está contraindicado en pacientes con feocromocitoma o con antecedentes de feocromocitoma.
Precauciones generales: Aunque poco frecuentes, se han reportado casos de reacciones alérgicas, incluyendo reacciones anafilácticas, erupción, edema angioneurótico y urticaria, en pacientes que estaban tomando atomoxetina. Muy raramente, se han notificado de forma espontánea casos de daño hepático, manifestado con un incremento en las enzimas hepáticas y la bilirrubina con ictericia. También muy raramente, se han notificado casos de daño hepático grave, incluyendo fallo hepático agudo. Debe interrumpirse el tratamiento con atomoxetina y no se debe reiniciar en pacientes con ictericia o evidencia, mediante pruebas de laboratorio, de daño hepático. Se puede presentar un aumento de la presión sanguínea y la frecuencia cardíaca en pacientes que reciben atomoxetina. Sin embargo, datos controlados y no controlados en TDAH muestran que aproximadamente 8-12% de niños y adolescentes, y 6-10% adultos, experimentan cambios más pronunciados en la frecuencia cardíaca (20 latidos por minuto o más) y presión sanguínea (15-20 mm Hg o más). Por lo que se recomienda medir la presión sanguínea y la frecuencia cardíaca antes de comenzar el tratamiento y durante el tratamiento con atomoxetina, en pacientes con hipertensión, taquicardia, o enfermedades cardiovasculares o enfermedades cerebrovasculares. La atomoxetina debe usarse con precaución en pacientes con prolongación del intervalo QT congénita o adquirida o en pacientes con antecedentes familiares de prolongación del intervalo QT. Igualmente, debe evitarse su uso con fármacos que prolongan el QT. La presencia de retención urinaria y retardo en el vaciamiento urinario, en algunos pacientes bajo tratamiento con atomoxetina, se recomienda su uso con precaución en pacientes con obstrucción de las vías urinarias. Atomoxetina no debe utilizarse en pacientes menores de seis años ya que la eficacia y seguridad no han sido establecidas en este grupo de edad. La aparición o exacerbación del comportamiento agresivo u hostilidad, en pacientes con trastornos por déficit de atención con hiperactividad (TDAH), bajo tratamiento con atomoxetina, por lo que se recomienda mantener vigilado al paciente. Se debe considerar la interrupción del tratamiento en pacientes con el surgimiento de manifestaciones suicidas emergentes o que puedan ser precursores del surgimiento de suicidio (p.ej., ansiedad, agitación, ataques de pánico, insomnio, irritabilidad, hostilidad, agresividad, impulsividad, acatisia, hipomanía, manía), en particular, si estas manifestaciones son severas o abruptas en su inicio, o no fueron parte de los síntomas que presenta el paciente. Se han notificado comportamientos suicidas (intentos de suicidio e ideas de suicidio) en pacientes tratados con atomoxetina. En ensayos doble ciego, los comportamientos suicidas fueron poco frecuentes, si bien se observaron con mayor frecuencia en niños y adolescentes tratados con atomoxetina, comparados con aquellos tratados con placebo, en los que no se produjeron tales acontecimientos. En ensayos doble ciego con adultos no hubo diferencias entre atomoxetina y placebo en la frecuencia de comportamiento suicida. En los pacientes que están siendo tratados de TDAH debe vigilarse cuidadosamente la aparición o el empeoramiento del comportamiento suicida. Atomoxetina puede provocar somnolencia, mareos o fatiga, por lo que se le debe recomendar al paciente tener precaución al conducir o utilizar maquinaria.
Restricciones de uso durante el embarazo y la lactancia: Atomoxetina se clasifica como categoría C de riesgo en el embarazo. Debido a la falta de estudios sobre el uso de atomoxetina en el embarazo, se recomienda utilizar atomoxetina solo si el beneficio potencial justifica el riesgo potencial para el feto. En estudios en animales (ratas), se observó que atomoxetina y/o sus metabolitos se excretaban en la leche, se desconoce si se excreta a través de la leche en humanos. Por lo tanto, atomoxetina debe ser usada con precaución en mujeres en periodo de lactancia.
Reacciones secundarias y adversas: 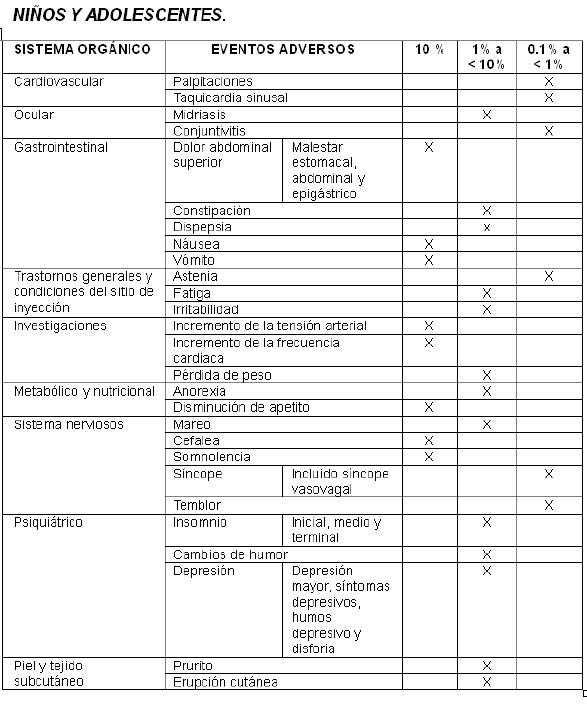
En pacientes metabolizadores lentos del CYP2D2 se observó, en menos del 2%, una frecuencia estadísticamente mayor de la presencia de los siguientes eventos adversos: Disminución de peso, constipación, insomnio, depresión, temblor, insomnio medio, síncope, conjuntivitis, despertar temprano por la mañana, midriasis, sedación. Pacientes pediátricos con trastorno por déficit de atención tratamos con atomoxetina experimentaron una disminución inicial en el promedio de ganancia de peso y talla.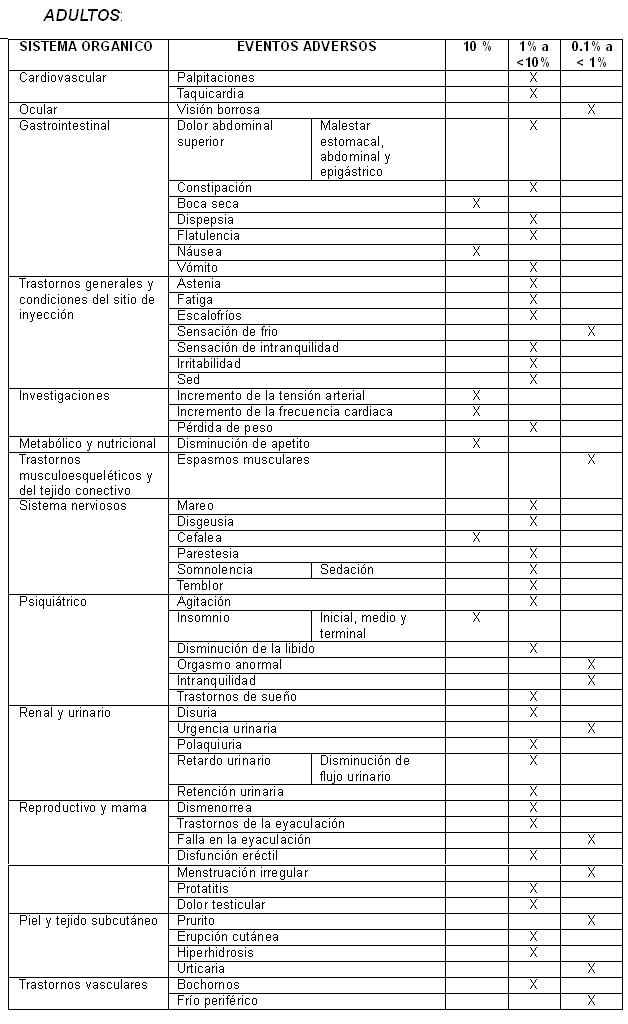
En pacientes metabolizadores lentos del CYP2D2 se observó, en menos del 2%, una frecuencia estadísticamente mayor de la presencia de los siguientes eventos adversos: Visión borrosa, boca seca, constipación, sensación de inquietud, disminución de apetito, temblor, insomnio, trastornos del sueño, insomnio medio, insomnio terminal, retención urinaria, disfunción eréctil, trastornos de la eyaculación, hiperhidrosis y frio periférico.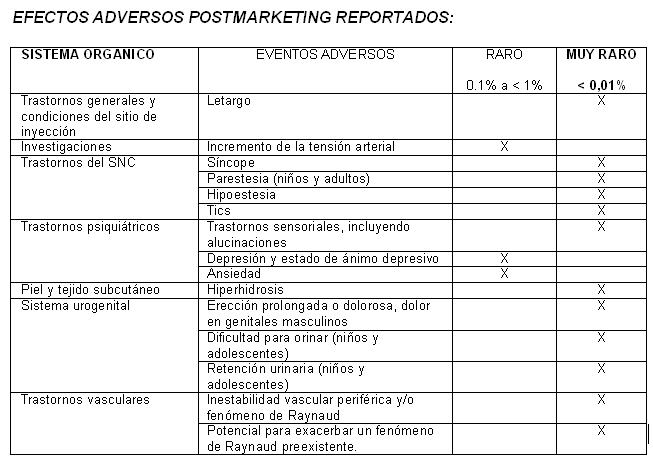
Interacciones medicamentosas y de otro género: Se aconseja precaución cuando se combine atomoxetina con inhibidores potentes de las enzimas del citocromo P450 distintas al CYP2D6 en pacientes que son metabolizadores lentos del CYP2D6 puesto que se desconoce el riesgo del aumento de las concentraciones plasmáticas de atomoxetina. Inhibidores de la enzima CYP2D6 como (fluoxetina, paroxetina, quinidina) pueden ocasionar un incremento de las concentraciones plasmáticas de la atomoxetina, se recomienda reajustar la dosis con la finalidad de evitar efectos tóxicos. Agonistas beta adrenérgicos (albuterol, salbutamol) se deben administrar con precaución a dosis altas mediante un nebulizador o por vía sistémica (oral o intravenosa) porque atomoxetina puede potenciar la acción de estos fármacos sobre el sistema cardiovascular. Atomoxetina debe emplearse con precaución con fármacos antihipertensivos, al posible aumento de la presión arterial, manteniendo en observación durante el tratamiento. Algunos medicamentos incluyendo antidepresivos (como imipramina, venlafaxina y mirtazapina) o descongestivos como (pseudoefedrina o fenilefrina), pueden actuar sobre la noradrenalina cuando se administren conjuntamente con atomoxetina, pudiendo ocasionar un efecto sinérgico o aditivo en su actividad farmacológica, deben utilizarse con precaución. La coadministración del metilfenidato y atomoxetina no presentan efectos relevantes en las funciones cardiovasculares. Los medicamentos que elevan el pH gástrico (hidróxido de magnesio/hidróxido de aluminio, omeprazol) no presenta efecto alguno sobre la biodisponibilidad de atomoxetina. Fármacos que se unen en gran proporción a las proteínas plasmáticas a las concentraciones terapéuticas. Warfarina, ácido acetilsalicílico, fenitoína o diazepam no afectaron la unión de atomoxetina a la albúmina humana. Igualmente, atomoxetina no afectó la unión de estos compuestos a la albúmina humana.
Alteraciones en los resultados de pruebas de laboratorio: No se tienen reportes de alteraciones significativas en los resultados de las pruebas de laboratorio
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: En los estudios realizados en animales no hubo evidencia de carcinogenicidad, mutagenicidad o alteraciones de la fertilidad.
Dosis y vía de administración: Oral. En niños y adolescentes con un peso < 70 Kg: La dosis diaria recomendada es de 0.5 mg/kg que puede incrementarse después de un mínimo de 3 días a la dosis total diaria de aproximadamente de 1.2 mg/kg, administrada ya sea como dosis única al día en las mañanas o en dosis dividida en la mañana y tarde/noche. Después de 2 a 4 semanas de tratamiento con atomoxetina, la dosis total diaria puede incrementarse a la dosis máxima de 1.8 mg/kg en pacientes que no hayan alcanzado una respuesta óptima. La dosis total máxima recomendada en niños y adolescentes de hasta 70 kg de peso corporal es de 1.8 mg/kg o 120 mg. En niños, adolescentes y adultos con un peso > 70 Kg: La dosis inicial recomendada es de 40 mg al día con incrementos al tercer día de tratamiento, hasta alcanzar el objetivo de 80 mg por día. Se puede administrar en una sola ingesta o 2 veces al día (mañana, tarde/noche). Después de un periodo de 2 a 4 semanas de tratamiento, la dosis puede incrementarse hasta un máximo de 120 mg en pacientes que no hayan alcanzado una respuesta óptima. Pacientes con insuficiencia hepática: La dosis debe ser ajustada con base en la tolerancia y respuesta clínica del paciente, ya que la depuración de atomoxetina se puede reducir. Pacientes con insuficiencia renal (fase terminal): La dosis debe ser ajustada con base en la tolerancia y respuesta clínica del paciente, dado que puede aumentar la hipertensión en esta población. El tratamiento con atomoxetina puede ser administrada con o sin alimentos, también se puede interrumpir el tratamiento sin necesidad de disminuir la dosis gradualmente.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: Los síntomas de una sobredosis de atomoxetina y que se reportaron con mayor frecuencia fueron somnolencia, agitación, hiperactividad, comportamiento anormal y síntomas gastrointestinales. Otros signos y síntomas importantes se derivan a la activación del S.N.C., presentando un aumento de la presión arterial, taquicardia, boca seca e midriasis, con intensidad de leve-moderada. En casos muy aislados de sobredosis se observaron una prolongación en el intervalo QT y convulsiones, igualmente casos de sobredosis fatal que involucra atomoxetina y por lo menos otro medicamento concomitante. No se cuenta con información específica sobre el tratamiento de sobredosis con atomoxetina. En el tratamiento de la sobredosis se recomienda tener una adecuada aireación como la vigilancia de los signos vitales y cardíacos junto con las medidas apropiadas sintomáticas y de soporte. El vaciado gástrico y la administración repetida de carbón activado (con/sin agentes catárticos) pueden evitar la absorción sistémica.
Presentación(es): Caja con 14 o 28 cápsulas con 25 mg. Caja con 14 o 28 cápsulas con 40 mg. Caja con 14 o 28 cápsulas con 60 mg.
Recomendaciones sobre almacenamiento: Consérvese la caja bien cerrada. Consérvese a no más de 30°C.
Leyendas de protección: Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. Reporte las sospechas de reacción adversa al correo farmacovigilancia@cofepris.gob.mx. Su uso durante el embarazo y la lactancia queda bajo la responsabilidad del médico. Este medicamento puede producir somnolencia y afectar el estado de alerta, por lo que no deberá conducir vehículos automotores ni maquinaria pesada durante su uso.
Nombre y domicilio del laboratorio: Hecho en México por: NEOLPHARMA, S. A. DE C. V. Blvd. de los Ferrocarriles No. 277, Col. Industrial Vallejo, C. P. 02300, Deleg. Azcapotzalco, D. F., México. Para: PSICOFARMA, S. A. DE C. V. Calz. Tlalpan No. 4369, Col. Toriello Guerra, C. P. 14050, Deleg. Tlalpan, D. F., México.
Número de registro del medicamento: 123M2015 SSA IV