METALYSE®
BOEHRINGER PM
Denominación genérica: Tenecteplasa.
Forma farmacéutica y formulación: Solución inyectable. El frasco ámpula con liofilizado contiene: tenecteplasa 50 mg (10.000 U). Excipiente cs. La jeringa con diluyente contiene: agua inyectable 10 ml. La solución reconstituida contiene 1.000 unidades (5 mg) de tenecteplasa por ml. La potencia de tenecteplasa es expresada en unidades (U) utilizando un estándar de referencia, el cual es específico para tenecteplasa y no es comparable con las unidades utilizadas para otros agentes trombolíticos.
Indicaciones terapéuticas: METALYSE® está indicado en el tratamiento trombolítico del infarto agudo al miocardio (IAM). El tratamiento debe ser iniciado tan pronto sea posible después del inicio de los síntomas.
Farmacocinética y farmacodinamia: La tenecteplasa es una proteína recombinante que activa el plasminógeno y se administra por vía intravenosa. Para ser eliminada de la circulación, se une a receptores específicos en el hígado y luego efectúa el catabolismo a péptidos pequeños. Sin embargo, su unión a receptores hepáticos es menor que la del t-PA natural, por lo que su vida media es más prolongada. En estudios en ratas con tenecteplasa marcada radioactivamente, se obtuvieron datos sobre la distribución tisular y la eliminación. El órgano principal en el que la tenecteplasa se distribuyó fue el hígado. Se desconoce si la tenecteplasa se une a las proteínas plasmáticas humanas y, de producirse, en qué se efectúa esta unión. Después de inyectar un bolo intravenoso único de tenecteplasa a pacientes con infarto agudo de miocardio, el antígeno tenecteplasa muestra una eliminación bifásica del plasma. En el rango de la dosis terapéutica, la depuración de tenecteplasa no es dependiente de la dosis. La vida media dominante inicial es de 24 ± 5,5 (media ± DE) min, cinco veces más prolongada que la del t-PA natural. La vida media terminal es de 129 ± 87 min y la depuración plasmática, de 119 ± 49 ml/min. El incremento del peso corporal llevó a que la depuración de tenecteplasa aumente en forma moderada y, a mayor edad, disminuyó ligeramente. Por lo general, las mujeres presentan menor depuración que los hombres, pero esto se puede deber a que en ellas el peso corporal suele ser inferior. Dado que la tenecteplasa se elimina por el hígado, no se espera que la disfunción renal afecte la farmacocinética de METALYSE®. Esto está fundamentado por datos en animales. Sin embargo, el efecto de la disfunción renal y hepática en la farmacocinética de la tenecteplasa en seres humanos no se investigó de manera específica. Tenecteplasa es un activador del plasminógeno tisular (t-PA) obtenido por tecnología recombinante del ADN, específico para fibrina, que se deriva del t-PA natural mediante modificaciones en tres lugares de la estructura proteica. Se une al componente de fibrina del trombo (coágulo de sangre) y selectivamente convierte el plasminógeno unido al trombo a plasmina, la que degrada la matriz del trombo. Tenecteplasa tiene una mayor especificidad para fibrina y mayor resistencia a la inactivación por su inhibidor endógeno (PAI-1) comparado con el t-PA natural. Luego de la administración de tenecteplasa se ha observado el consumo dosis-dependiente de alfa2-antiplasmina (el inhibidor de plasmina en fase líquida) con el consecuente aumento en el nivel de generación sistémica de plasmina. Esta observación es consistente con el efecto pensado de la activación del plasminógeno. En estudios comparativos una reducción de menos de 15% en fibrinógeno y una reducción de menos de 25% en plasminógeno fueron observadas en sujetos tratados con la máxima dosis de tenecteplasa (10.000 U, correspondiente a 50 mg), mientras que alteplasa causó aproximadamente una disminución de 50% en los niveles de fibrinógeno y plasminógeno. No se detectó formación de anticuerpos clínicamente relevante a 30 días. Datos de dilatación de los estudios angiográficos de fase I y II sugieren que tenecteplasa, administrado como un bolo intravenoso único, es efectivo en disolver los coágulos sanguíneos en la arteria relacionada con el infarto de sujetos experimentando un IAM en una forma relacionada a la dosis. Un ensayo de mortalidad a gran escala (ASSENT 2) en aproximadamente 17.000 pacientes mostró que tenecteplasa es terapéuticamente equivalente a alteplasa en reducir la mortalidad (6,2% para ambos tratamientos, a 30 días) y que el uso de tenecteplasa está asociado con una incidencia significativamente menor de sangrado no intracraneal (26,4% vs. 28,9%, p=0,0003). La disminución del riesgo de hemorragias probablemente se relacione con que la tenecteplasa presenta mayor especificidad para la fibrina y supone un régimen adaptado al peso. Esto se traduce en una necesidad significativamente menor de transfusiones (4,3% vs. 5,5%, p=0,0002). La hemorragia intracraneal ocurrió en una tasa de 0,93% vs. 0,94% para tenecteplasa y alteplasa, respectivamente. En los 475 pacientes tratados después de 6 horas se observaron diferencias numéricas a favor de tenecteplasa con respecto a la mortalidad a 30 días (4,3% vs. 9,6%), accidente cerebrovascular (0,4% vs. 3,3%) y HIC (0% vs. 1,7%). El objetivo del estudio ASSENT 3 fue optimizar la terapia antitrombótica concomitante con tenecteplasa a fin de mejorar las tasas de permeabilidad temprana y mantener la perfusión, sobre todo para superar el efecto paradójico procoagulante que se produce por la liberación de la trombina atrapada debido a la lisis del coágulo. Se compararon tres regímenes antitrombóticos concomitantes diferentes en 6095 pacientes: dosis completa de tenecteplasa + heparina no fraccionada (HNF) vs. dosis completa de tenecteplasa + heparina de bajo peso molecular (BPM) (enoxaparina) vs. media dosis de tenecteplasa + heparina no fraccionada + dosis completa de abciximab. La HNF se administró de acuerdo con las pautas de la AHA/ACC con un régimen completo de baja dosis adaptado al peso corporal: un bolo único IV de 60 UI/kg (máximo, 4.000 UI) seguido de inmediato por una infusión IV de 12 UI/kg/h (máximo, 1.000 UI/H) durante las tres primeras horas y a partir de allí, según el tiempo de tromboplastina parcial activado (TTPA), con monitoreo por 48 horas para mantener el TTPA en 50-70 segundos. La tasa de mortalidad a los 30 días fue, respectivamente, 6,0%, 5,4% y 6,6%; la de hemorragias mayores intrahospitalarias (salvo HIC), 2,16%, 3,04% y 4,32%; y la de HIC, 0,93%, 0,88% y 0,94%. En ASSENT 3, el régimen completo de HNF en baja dosis adaptado al peso corporal que recomienda la AHA/ACC se administró en forma concomitante con tenecteplasa y causó menos hemorragias sistémicas pero tasas de HIC similares a las del régimen de dosificación de HNF más intensivo que se suministró en ASSENT 2, sin pérdida de la eficacia. ASSENT 3 PLUS, un estudio satélite de ASSENT 3, se diseñó para investigar el contexto prehospitalario. La eficacia y la seguridad de la dosis completa de tenecteplasa + HNF vs. la dosis completa de tenecteplasa + heparina de BPM (enoxaparina) se evaluaron en 1639 pacientes. El diseño del estudio y las dosificaciones de los tratamientos fueron idénticos a los de ASSENT 3. La terapia de reperfusión prehospitalaria con tenecteplasa y HNF o enoxaparina permitió tratar a > 50% de los pacientes con infarto de miocardio con elevación de ST (IMEST) dentro de las dos horas del comienzo de los síntomas. En los estudios ASSENT 3 y 3 PLUS, la terapia adyuvante prehospitalaria e intrahospitalaria con enoxaparina disminuyó la incidencia de las complicaciones isquémicas frente a la terapia adyuvante con HNF: la incidencia del criterio de valoración de la eficacia a los 30 días combinado (muerte, reinfarto, isquemia refractaria) fue, respectivamente, 11,4% vs. 15,4 en ASSENT 3 y 14,2% vs. 17,4% en ASSENT 3 PLUS. Sin embargo, en el contexto prehospitalario, la tenecteplasa con la enoxaparina a las dosis administradas se asoció con mayor riesgo de hemorragia mayor y HIC en pacientes > 75 años. La permeabilidad coronaria y los datos limitados de los resultados clínicos mostraron que los pacientes con IAM fueron tratados satisfactoriamente seis horas después de la aparición de los síntomas. El estudio ASSENT-4 PCI se diseñó para evaluar si en 4.000 pacientes con infarto de miocardio extenso el pretratamiento con dosis completas de tenecteplasa y un bolo único concomitante de hasta 4000 UI de heparina no fraccionada, administrados antes de una intervención coronaria percutánea (ICP) primaria a realizar dentro de los 60-180 minutos posteriores, proporciona mejores resultados que la ICP primaria sola. El estudio se detuvo prematuramente cuando 1.667 pacientes habían sido asignados aleatoriamente, debido a la mortalidad numéricamente mayor en el grupo de la ICP facilitada que recibía tenecteplasa. La incidencia del criterio de valoración principal, una combinación de muerte o shock cardiogénico o insuficiencia cardíaca congestiva dentro de los 90 días, fue significativamente mayor en el grupo tratado con el régimen exploratorio de tenecteplasa seguida de ICP inmediata de rutina: 18,6% (151/810) frente a 13,4% (110/819) en el grupo en el que sólo se efectuó ICP, p=0,0045. Esta diferencia significativa entre los grupos en cuanto al criterio de valoración primario a los 90 días ya se había observado en el contexto intrahospitalario y a los 30 días. Numéricamente, todos los componentes del criterio de valoración clínica principal combinado fueron favorables para el tratamiento con ICP sola: muerte: 6,7% vs. 4,9%, p=0,14; shock cardiogénico: 6,3% vs. 4,8%, p=0,19; insuficiencia cardíaca congestiva: 12,0% vs. 9,2%, p=0,06, respectivamente. Los criterios de valoración secundarios, reinfarto y revascularización repetida de los vasos diana, aumentaron significativamente en el grupo tratado previamente con tenecteplasa: reinfarto: 6,1% vs. 3,7%, p=0,0279; revascularización repetida de los vasos diana: 6,6% vs 3,4%, p=0,0041. Los siguientes eventos adversos se presentaron con más frecuencia al administrar tenectaplasa antes de la ICP: hemorragia intracraneal: 1% vs. 0%, p=0,0037; accidente cerebrovascular: 1,8% vs. 0%, p < 0,0001; hemorragias mayores: 5,6% vs. 4,4%, p=0,3118; hemorragias menores: 25,3% vs. 19,0%, p=0,0021; transfusiones de sangre: 6,2% vs. 4,2%, p=0,0873; cierre brusco del vaso: 1,9% vs. 0,1%, p=0,0001.
Contraindicaciones: La terapia trombótica se asocia con riesgo de hemorragia. METALYSE® está contraindicado en las siguientes situaciones: trastorno hemorrágico significativo actual o dentro de los últimos seis meses, diátesis hemorrágica conocida; pacientes con terapia anticoagulante oral concomitante en el presente (cociente internacional normalizado INR > 1,3); antecedentes de lesión del sistema nervioso central (ej., neoplasma, aneurisma, cirugía intracraneal o espinal); hipertensión grave, no controlada; cirugía mayor, biopsia de un órgano parenquimatoso o traumatismo significativo en los últimos dos meses (incluye cualquier traumatismo asociado con el IAM actual), traumatismo craneoencefálico reciente; reanimación cardiopulmonar prolongada o traumática ( > 2 minutos) dentro de las dos últimas semanas; disfunción hepática grave, incluidas insuficiencia hepática, cirrosis, hipertensión portal (várices esofágicas) y hepatitis activa; úlcera péptica activa; aneurisma arterial y malformación arterial/venosa conocida; neoplasma con aumento del riesgo de hemorragia; pericarditits aguda y/o endocarditis bacterial subaguda; pancreatitis aguda; hipersensibilidad al principio activo tenecteplasa, gentamicina (trazas de residuos del proceso de manufactura) o a cualquiera de los excipientes; accidente cerebrovascular hemorrágico o de origen desconocido en cualquier momento; accidente cerebrovascular isquémico o accidente isquémico transitorio (AIT) en los últimos seis meses.
Precauciones generales: METALYSE® debe ser prescrito por médicos con experiencia en el uso de tratamiento trombolítico y con facilidades para monitorizar su uso. Esto no impide el uso pre-hospitalario de METALYSE®. Como con otros trombolíticos, se recomienda que cuando METALYSE® sea administrado, un equipo de resucitación estándar y de medicación se encuentre disponible en todas las circunstancias. Intervención coronaria percutánea (ICP) primaria: si se programa una ICP primaria de acuerdo con las pautas de tratamiento relevantes en la actualidad, METALYSE®, como se administró en el estudio de ICP ASSENT-4, no se debe suministrar. Sangrado: la complicación más común encontrada con la terapia de METALYSE® es el sangrado. El uso concomitante de anticoagulación con heparina puede contribuir al sangrado. Ya que la fibrina es destruida durante la terapia con METALYSE®, puede ocurrir sangrado de lugares de punciones recientes. Por lo tanto, la terapia trombolítica requiere atención cuidadosa a todo lugar de sangrado posible (incluyendo aquellos luego de la inserción de catéter, punción arterial y venosa, cortes y punción de aguja). El uso de catéteres rígidos, inyecciones intramusculares y manipulación no esencial del paciente deben ser evitados durante el tratamiento con METALYSE®. Si ocurriera sangrado serio, hemorragia cerebral en particular, la administración de heparina concomitante debe ser terminada inmediatamente. Se debe considerar la administración de protamina si la heparina ha sido administrada en las 4 horas previas al inicio del sangrado. En los pocos pacientes que fallan en responder a estas medidas conservadoras, el uso sensato de productos de transfusión puede estar indicado. La transfusión de crioprecipitado, plasma fresco congelado y plaquetas debe ser considerada con una reevaluación clínica y de laboratorio luego de cada administración. Es deseable el objetivo de un nivel de fibrinógeno de 1 g/l con la infusión de crioprecipitado. Los agentes antifibrinolíticos deben ser también considerados. El uso de la terapia de METALYSE® tiene que ser cuidadosamente evaluado para balancear el potencial riesgo de sangrado con los beneficios esperados bajo las siguientes condiciones: presión arterial sistólica > 160 mm/Hg; enfermedad cerebrovascular; sangrado gastrointestinal o genitourinario reciente (en los últimos 10 días); cualquier inyección intramuscular reciente conocida (en los últimos 2 días); edad avanzada, por ejemplo mayor de 75 años; bajo peso corporal < 60 kg; parto reciente. Arritmias: la trombolisis coronaria puede resultar en arritmia asociada con la reperfusión. Las arritmias re-perfundidas pueden conducir a un paro cardíaco, poner en peligro la vida y requerir el uso de terapias antiarrítmicas convencionales. Antagonistas de la glicoproteína IIb/IIIa: el uso concomitante de antagonistas de GPIIb/IIIa incrementa el riesgo de sangrado. Tromboembolismo: el uso de METALYSE® puede aumentar el riesgo de eventos trombóticos en pacientes con trombos en el corazón izquierdo, ej., estenosis mitral o fibrilación atrial. Hipersensibilidad: después del tratamiento no se ha observado formación de anticuerpos a la molécula de tenecteplasa. Sin embargo, no existe experiencia con la re-administración de METALYSE®. Son raras las reacciones anafilactoides asociadas con la administración de METALYSE® y pueden ser ocasionadas por hipersensibilidad al principio activo tenecteplasa, gentamicina (trazas de residuos del proceso de manufactura) o a cualquier excipiente. Si ocurre una reacción anafilactoide, debe ser descontinuada la administración e iniciarse el tratamiento apropiado.
Restricciones de uso durante el embarazo y la lactancia: No existe experiencia alguna del uso de METALYSE® en el embarazo. El beneficio deberá ser evaluado contra el riesgo potencial en caso de infarto de miocardio durante embarazo. No se sabe si tenecteplasa es excretado en la leche materna.
Reacciones secundarias y adversas: Así como con otros agentes trombolíticos, la hemorragia es el efecto más común no deseable asociado con el uso de METALYSE®. Se puede producir hemorragia en cualquier sitio o cavidad corporal, y puede provocar situaciones potencialmente fatales, discapacidad permanente o muerte. El tipo de hemorragia asociado con terapia trombolítica puede ser dividida en dos grandes categorías: 1. Sangrado superficial, normalmente en sitios de inyección. 2. Sangrados internos en cualquier sitio o cavidad corporal. Los síntomas neurológicos como somnolencia, afasia, hemiparesia y convulsiones pueden estar asociados a hemorragia intracraneal. Trastornos del sistema inmune: reacciones anafilactoides (rash, urticaria, broncoespasmo, edema laríngeo, angioedema). Trastornos del sistema nervioso: hemorragia intracraneal (como hemorragia cerebral, hematoma cerebral, accidente cerebro-vascular hemorrágico, transformación hemorrágica del accidente cerebro-vascular, hematoma intracraneal, hemorragia subaracnoidea). Trastornos oculares: hemorragia ocular. Trastornos cardíacos: arritmias de reperfusión (asistolia, arritmia idioventricular acelerada, arritmia, extrasístoles, fibrilación auricular, bloqueo auriculoventricular de primer grado, bloqueo auriculoventricular completo, bradicardia, taquicardia, arritmia ventricular, fibrilación ventricular, taquicardia ventricular) ocurren en una muy cercana relación temporal bajo tratamiento con METALYSE®. Las arritmias de reperfusión pueden producir un paro cardíaco, que comprometa la vida y puede requerir el uso de terapias antiarritmicas convencionales. Hemorragia pericárdica. Trastornos vasculares: hemorragia. Embolismo. Trastornos respiratorios, torácicos y mediastinales: epistaxis. Hemorragia pulmonar. Trastornos gastrointestinales: hemorragia gastrointestinal (hemorragia gástrica, úlcera gástrica y hemorragia, hemorragia rectal, hematemesis, melena, hemorragia bucal), náuseas, vómitos. Hemorragia retroperitoneal (como hematoma retroperitoneal). Trastornos de piel y tejido celular subcutáneo: equimosis. Trastornos renales y urinarios: hemorragia urogenital (como hematuria, hemorragia del tracto urinario). Trastornos generales y condiciones del sitio de administración: hemorragia en el sitio de inyección, hemorragia en el sitio de punción. Investigaciones: disminución de la presión arterial. Incremento de la temperatura corporal. Daño, envenenamiento y complicaciones del procedimiento: embolismo graso que puede tener consecuencias en los órganos afectados. Procedimientos médicos y quirúrgicos: transfusión.
Interacciones medicamentosas y de otro género: No se ha desarrollado ningún estudio formal de interacciones con METALYSE® respecto a productos farmacéuticos comúnmente administrados en pacientes con presencia de IAM; sin embargo, el análisis de los datos de más de 12.000 pacientes tratados durante las fases I, II y III del desarrollo del medicamento no reveló ninguna interacción clínicamente relevante con los medicamentos comúnmente utilizados en pacientes con IAM. Los medicamentos que alteran la coagulación o la función plaquetaria, pueden aumentar el riesgo de sangrado antes, durante o después de la terapia con METALYSE®. METALYSE® no es compatible con la solución de dextrosa. No se debe de adicionar otro medicamento a la solución de METALYSE® o a la línea de infusión cuando se esté administrando éste.
Alteraciones en los resultados de pruebas de laboratorio: Los resultados de las pruebas de laboratorio relacionadas con los tiempos de sangrado y de coagulación pueden sufrir modificación durante el período de aplicación de METALYSE® tal como sucede con la terapia trombolítica y fibrinolítica en general. La administración de dosis única intravenosa en ratas, conejos y perros resultó en alteraciones dosis-dependientes y reversibles de los parámetros de coagulación con hemorragia local en el sitio de la inyección, que fue atribuido como consecuencia del efecto farmacodinámico de la tenecteplasa. Estudios de toxicidad con dosis múltiple en ratas y perros confirmaron las observaciones mencionadas previamente, pero la duración del estudio estuvo limitada a dos semanas por formación de anticuerpos a la proteína humana tenecteplasa, que resultó en anafilaxia. Datos farmacológicos de seguridad en monos cinomolgos reveló reducción de la tensión arterial seguida de cambios transitorios en el electrocardiograma pero esto ocurrió en exposiciones que fueron considerablemente mayores a la exposición clínica.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: En relación a la indicación y la administración de dosis única en humanos, la evaluación de toxicidad reproductiva fue reducida a los conejos, como especie sensible. La tenecteplasa no indujo teratogenicidad. La administración repetida resultó en sangrado con mortalidad secundaria en hembras madres. En algunos casos ocurrió aborto y reabsorción del feto. No se observaron efectos después de la administración de dosis única de tenecteplasa. No se espera mutagenicidad o carcinogenicidad para esta clase de proteínas recombinantes y las evaluaciones de genotoxicidad y carcinogenicidad no fueron necesarias. No se observó irritación local de los vasos sanguíneos después de la administración intravenosa, intra-arterial o para venosa de la formulación final de tenecteplasa.
Dosis y vía de administración: Intravenosa por bolo. METALYSE® debe ser administrado en base al peso corporal, con un máximo de dosificación de 10.000 UI (50 mg de tenecteplasa). El volumen requerido para administrar la dosis correcta puede ser calculado en base al siguiente esquema: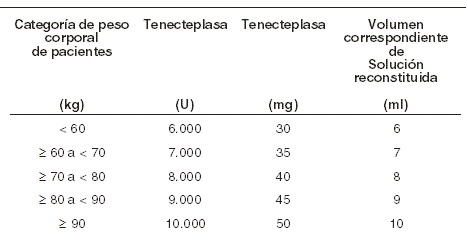
La dosis requerida debe ser administrada en bolo intravenoso único en un lapso de aproximadamente 5-10 segundos. Puede utilizarse una línea venosa preexistente para la administración de METALYSE®, la cual podrá seguir siendo utilizada exclusivamente para la administración de solución sódica al 0,9%. METALYSE® no es compatible con la solución de dextrosa. Si se desea continuar utilizando dicha línea se deberá "enjuagar" esta línea tras la administración de METALYSE® para una administración continua adecuada. Ningún otro medicamento debe ser agregado a la solución de METALYSE® (ni siquiera heparina). Terapia concomitante: la terapia antitrombótica adjunta se recomienda de acuerdo a las guías internacionales actuales para el manejo de pacientes con infarto agudo de miocardio con elevación del segmento ST. Instrucciones de uso:

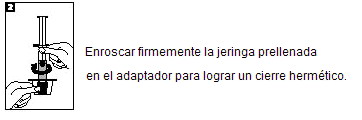





METALYSE® debe ser reconstituido añadiendo el volumen completo de agua para inyectables de la jeringa precargada al vial conteniendo el polvo para inyección. Asegúrese que el tamaño del vial sea seleccionado acorde al peso corporal del paciente (vea Dosis y vía de administración). Revise que la tapa del vial esté aún intacta. Retire la tapa del vial. Retire la tapa de la jeringa. Luego atornille inmediatamente la jeringa prellenada en el adaptador del frasco y penetre el tapón del frasco en el centro con el punto del adaptador del mismo. Agregue el agua para inyectables al vial empujando el émbolo de la jeringa lentamente hacia abajo para evitar hacer espuma. Reconstituir girándolo suavemente. La preparación reconstituida es una solución transparente entre incolora y amarillo pálido. Sólo se debe administrar una solución sin partículas. Inmediatamente antes de la administración de la solución, invierta el vial con la jeringa aún ligada, de tal manera que la jeringa está debajo del vial. Saque el volumen apropiado de solución reconstituida de METALYSE® a la jeringa, basado en el peso del paciente. Desconecte la jeringa del vial. METALYSE® debe ser administrado al paciente por vía intravenosa en aproximadamente 5 a 10 segundos. No debe ser administrado a la vía conteniendo dextrosa. Cualquier solución no utilizada debe ser descartada. Alternativamente la reconstitución puede ser realizada con la aguja incluida.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: En caso de sobredosificación puede incrementarse el riesgo de sangrado. En caso de sangrado prolongado deberá de considerarse la sustitución de la terapia.
Presentación(es): Frascos-ámpula con 50 mg (10.000 U) de liofilizado de tenecteplasa y jeringa prellenada con 10 ml de agua inyectable para reconstituir la solución.
Recomendaciones sobre almacenamiento: Consérvese a temperatura ambiente no mayor de 30°C. Protéjase de la luz.
Leyendas de protección: Dosis: la que el médico señale. Léase instructivo. Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. No se use en el embarazo ni en la lactancia.
Nombre y domicilio del laboratorio: Hecho en Alemania por: Boehringer Ingelheim Pharma GmbH & Co., KG Birkendorferstrasse 65, D-88397Biberach an der Riss, Alemania. Distribuido por: Boehringer Ingelheim Promeco S.A. de C.V., Calle del Maíz No. 49, Barrio Xaltocan, Xochimilco.16090, México, D.F. ®Marca registrada.
Número de registro del medicamento: 449M2001 SSA IV.
Clave de IPPA: 083300RR011017