NAGLAZYME®
STENDHAL
Denominación genérica: Galsulfasa.
Forma farmacéutica y formulación: Solución. El frasco ámpula de 5ml contiene: galsulfasa 5 mg. Vehículo cbp 5 ml. NAGLAZYME® contiene los siguientes excipientes en su formulación: cloruro de sodio, fosfato de sodio monobásico monohidratado, fosfato de sodio dibásico heptahidratado, polisorbato 80 y agua para preparaciones inyectables.
Indicaciones terapéuticas: NAGLAZYME® está indicado para el tratamiento enzimático sustitutivo a largo plazo en pacientes con diagnósticos confirmado de mucopolisacaridosis VI (MPS VI, deficiencia de N-acetilgalactosamina 4-sulfatasa; síndrome de Maroteaux-Lamy). Como sucede con todas la enfermedades lisosomales hereditarias, es de suma importancia, especialmente en las formas graves, iniciar el tratamiento tan pronto como sea posible, antes de la aparición de las manifestaciones clínicas irreversibles de la enfermedad. Es fundamental inicial el tratamiento en pacientes menores de 5 años con una forma grave de la enfermedad, a pesar de que no se incluyera a pacientes menores de 5 años en el ensayo clínico de fase 3. Grupo farmacoterapéutico: otros preparados para el aparato digestivo y metabolismo enzimas, código ATC: A16AB. Los tres ensayos clínicos son NAGLAZYME® se centraron en la evaluación de las manifestaciones sistémicas de la MPS VI como resistencia, movilidad articular, dolor y rigidez articulares, obstrucción respiratoria superior, destreza manual y agudeza visual. La seguridad y eficacia de NAGLAZYME® se evaluaron en un ensayo en fase 3, aleatorio, doble ciego, controlado con placebo en 39 pacientes con MPS VI, de 5 a 29 años de edad. La mayoría de los paciente presentaba baja estatura, deterioro de la resistencia y síntomas musculoesqueléticos. Se incluyó en el ensayo de los pacientes que al inicio del mismo podían andar más de 5 m pero menos de 250 m en 6 minutos, o menos de 400 m en la prueba de la caminata de 12 minutos (12 Minute Walk Test). Los pacientes recibieron 1 mg/kg de galsulfasa o placebo cada semana durante un total de 24 semanas. La variable principal de eficacia fue el número de metros caminados en 12 minutos en la semana 24, en comparación con el número de metros caminados al inicio del ensayo. Las variables secundarias de eficacia fueron el número de peldaños por minuto de 3 minutos en la prueba de subir escaleras y la excreción urinaria de glucosaminoglucanos en pacientes tratados con NAGLAZYME® en comparación con placebo en la semana 24. Posteriormente, 38 pacientes participaron en una extensión abierta del ensayo en la que recibieron 1 mg/kg de galsulfasa cada semana. Tras 24 de semanas de tratamiento, los pacientes que recibieron NAGLAZYME® experimentaron una mejoría de 92 ± 40 m de la distancia caminada en 12 minutos respecto con los pacientes tratados con placebo (p=0,025). Los pacientes tratados experimentaron una mejoría de 5,7 peldaños por minuto en la prueba de subir escaleras en 3 minutos (3 Minute Stair Climb) respecto de los pacientes tratados con placebo. Los paciente tratados con NAGLAZYME® experimentaron asimismo una reducción de la excreción urinaria de glucosaminoglucanos de 238 ± 17,8 mg/mg de creatinina (media ± DE) tras 24 semanas de tratamiento respecto de los pacientes tratados con placebo. Los resultados de la determinación de GAG se aproximaron al intervalo normal para el grupo de edad en los pacientes tratados con NAGLAZYME®.
Farmacocinética y farmacodinamia: Se evaluó la farmacocinética de galsulfasa en 13 pacientes con MPS VI que recibieron 1 mg/kg de galsulfasa en una infusión de 4 horas. Tras 24 semanas de tratamiento, la media de la concentración plasmática máxima (Cmáx.) fue de 2,357 (± desviación estándar [SD] 1,560) ng/ml y la del área bajo la curva (AUC0-t) fue de 5,860 (±SD 4,184) h x ng/ml. El volumen de distribución (Vz) medio fue de 316 (±SD 752) ml/kg y el aclaramiento plasmático (CL) medio fue de 7,9 (± 14,7) ml/min/kg. La semivida de eliminación (t1/2) media fue de 22,8 (± 10,7) minutos en la semana 24. Los parámetros farmacocinéticos obtenidos de pacientes en ensayos de fase 1 se han mantenido estables a largo plazo (al menos hasta 194 semanas). La galsulfasa es una proteína y cabe esperar su catabolismo por hidrólisis peptídico. Por consiguiente, la insuficiencia hepática no debería alterar la farmacocinética de galsulfatasa de manera clínicamente significativa. El aclaramiento renal de galsulfasa se considera una vía minoritaria de eliminación. Las enfermedades por almacenamiento de mucopolisacáridos están ocasionadas por la deficiencia de enzimas lisosómicas específicas necesarias para el catabolismo de los glucosaminoglucanos (GAG). La MPS VI es un trastorno heterogéneo y multisistémico caracterizado por la deficiencia de N-acetilgalactosamina 4-sulfatasa, una hidrolasa lisosómica que cataliza la hidrólisis de los grupos sulfato de un glucosaminoglucano, el dermatán sulfato. La reducción o ausencia de la actividad de N-acetilgalactosamina 4-sulfatasa tiene como resultado la acumulación de dermatán sulfato en muchos tipos celulares y tejidos. El fundamento del tratamiento enzimático sustitutivo es establecer un nivel de actividad enzimática suficiente para hidrolizar el sustrato acumulado y evitar su acumulación. La galsulfasa purificada, una forma recombinante de la N-acetilgalactosamina 4-sulfatasa humana, es una glucoproteína con un peso molecular aproximado de 56 kD. La galsulfasa contiene 495 aminoácidos, tras la escisión del extremo N-terminal. La molécula contiene 6 puntos de N-glicosilación con oligosacáridos. Tras la infusión intravenosa, la galsulfasa se elimina rápidamente de la circulación y es captada por las células, probablemente por los receptores manosa-6-fosfato, y transportadas a los lisomas.
Contraindicaciones: Hipersensibilidad al principio activo o a alguno de los excipientes.
Precauciones generales: Se recomienda precaución en el manejo y tratamiento de pacientes con afectación de las vías aéreas, limitando o monitorizando rigurosamente el uso de antihistamínicos y de medicamentos sedantes. Asimismo, se considerará recurrir a la presión positiva de las vías aéreas durante el sueño y a una posible traqueotomía en situaciones clínicas que lo requieran. Debe valorarse la posibilidad de retrasar la infusión de NAGLAZYME® en pacientes que presenten una enfermedad febril o respiratoria aguda. No se ha establecido la seguridad y la eficacia de NAGLAZYME® en niños menores de 5 años ni en pacientes mayores de 65 años. No se ha evaluado la seguridad y la eficacia de NAGLAZYME® en pacientes con insuficiencia renal o epática. Los pacientes tratados con NAGLAZYME® han desarrollado reacciones asociadas a la infusión (RAI) definidas como cualquier reacción adversa relacionada con el medicamento durante la infusión o hasta el final del día de la infusión. En los ensayos clínicos con NAGLAZYME®, las RAI habitualmente respondieron a la interrupción o reducción del ritmo de la infusión y pretratando al paciente con antihistamínicos y/o antipiréticos (paracemol), lo que permite que el paciente continúe el tratamiento. Como se dispone de experiencia limitada con la reanudación del tratamiento tras una interrupción prolongada, se requiere precaución debido al incremento del riesgo teórico de una reacción de hipersensibilidad. En el tratamiento con NAGLAZYME®, se recomienda la administración de un pretratamiento farmacológico (antihistamínicos con o sin antipiréticos) aproximadamente 30-60 minutos antes del inicio de la infusión para minimizar la aparición de posibles RAI. En caso de RAI leve o moderada, se considerará el tratamiento con antihistamínicos y paracetamol y/o la reducción de la velocidad de infusión a la mitad de la velocidad a la que se produjo la reacción. En caso de una única RAI grave, deberá detenerse la infusión hasta la resolución de los síntomas y se considerará el tratamiento con antihistamínicos y paracetamol. La infusión se reiniciará con una reducción de la velocidad de infusión al 50%-25% de la velocidad a la que se produjo la reacción. En caso de una RAI moderada repetida o una reexposición tras una única RAI grave, se considerará el pretratamiento (antihistamínicos y paracetamol y/o conrtocosteroides) y una reducción de la velocidad de infusión al 50%-25% de la velocidad a la que se produjo la reacción. Como sucede con cualquier otro producto proteico de administración intravenosa, pueden aparecer reacciones de hipersensibilidad graves de tipo alérgico. Si se producen dichas reacciones, se recomienda la inmediata interrupción de NAGLAZYME® y la instauración del tratamiento médico adecuado. Deben considerarse los estándares médicos actuales en el tratamiento de emergencia. No se han realizado estudios sobre la capacidad para conducir y utilizar máquinas en pacientes que reciben NAGLAZYME®.
Restricciones de uso durante el embarazo y la lactancia: No existen datos clínicos sobre la utilización de NAGLAZYME® en mujeres embarazadas. Los estudios en animales no muestran efectos dañinos directos o indirectos sobre el embarazo o desarrollo embrional/fetal. NAGLAZYME® no debe utilizarse durante el embarazo, excepto si fuese claramente necesario. Se desconoce si la galsulfasa se excreta a la leche, por lo que debe interrumpirse la lactancia durante el tratamiento con NAGLAZYME®.
Reacciones secundarias y adversas: Todos los pacientes en el ensayo clínico de fase 3 experimentaron reacciones adversas medicamentosas (RAM), independientemente de que recibieran NAGLAZYME® o placebo. Las RAM notificadas durante el ensayo de fase 3, en 39 pacientes tratados durante 6 meses, se muestran en la tabla 1, clasificados por órganos y sistemas, siguiendo la convención recomendada sobre frecuencia. Las reacciones adversas muy frecuentes son las que aparecieron con una frecuencia superior a 1 por 10. Las reacciones adversas frecuentes aparecieron con una frecuencia superior a 1 por 100. Se produjo un caso de apnea del sueño durante este ensayo. Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada intervalo de frecuencia.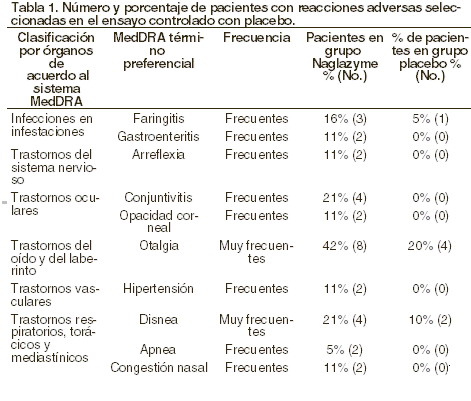
Las reacciones asociadas a la infusión, caracterizadas por un patrón recurrente de síntomas que aparecen durante la infusión de NAGLAZYME®, se observaron en 30 (56%) de los 54 pacientes tratados con NAGLAZYME® en todos los ensayos clínicos. Las reacciones se produjeron desde la semana y hasta la 55 del tratamiento con el fármaco de estudio, y durante múltiples infusiones, aunque no siempre en semanas consecutivas. Los síntomas más frecuentes de estas reacciones asociadas a la infusión fueron fiebre, escalofríos/temblores, exantema y urticaria, aunque también se han descrito hipotensión, náuseas, vómito, disnea, broncoespasmo, dolor retroesternal, dolor abdominal, cefalea, malestar general, estrés respiratorio, edema angioneurótico y artralgia.
Interacciones medicamentosas y de otro género: No se han realizado estudios de interacciones.
Alteraciones en los resultados de pruebas de laboratorio: Presentaron resultados positivos de anticuerpos IgG contra galsulfasa 53/54 pacientes (98%) tratados con NAGLAZYME® en los ensayos. La evidencia inicial de desarrollo de anticuerpos aparece típicamente tras 4 a 8 semanas de tratamiento. No se observó alguna relación entre la aparición de anticuerpos contra galsulfasa y los niveles urinarios de glucosaminoglicanos (GAG). Se desconoce el significado clínico de los anticuerpos contra galsulfasa.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad de dosis única, toxicidad de dosis repetidas o sobre toxicidad para la reproducción o el desarrollo embrionario y fetal en ratas o conejos. No se ha investigado la toxicidad perinatal y postnatal. No cabe esperar potencial genotóxico ni carcinogénico. Se desconoce la causa de la relevancia clínica de la toxicidad hepática (hiperplasia ductal biliar/inflamación periportal) observada en el estudio de dosis repetidas en monos con dosis clínicamente relevantes.
Dosis y vía de administración: Un médico experto en el tratamiento de pacientes con MPS VI u otras enfermedades metabólicas hereditarias debe supervisar el tratamiento con NAGLAZYME®. NAGLAZYME® se debe administrar en las instalaciones clínicas adecuadas en las que se disponga de acceso inmediato a un equipo de reanimación para el tratamiento de emergencias médicas. El régimen posológico recomendado de galsulfasa es de 1 mg/kg de peso corporal, administrada una vez a la semana como infusión intravenosa durante 4 horas. La velocidad inicial de infusión se debe ajustar de modo que durante la primera hora se administre aproximadamente un 2,5% del total de la solución y el volumen restante (aproximadamente el 97,5%) durante las siguientes 3 horas. Cada vial de NAGLAZYME® es para un único uso. El contenido para solución para infusión debe diluirse con una solución de cloruro sódico al 0,9% para infusión mediante una técnica aséptica. Se recomienda que la solución diluida de NAGLAZYME® se administre a los pacientes mediante un equipo de infusión equipado con un filtro en línea de 0,2 micras. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con las normativas locales. Preparación de la infusión de NAGLAZYME® (se utilizará una técnica aséptica): se determinará el número de viales por diluir de acuerdo con el peso del paciente y se retirarán del refrigerador aproximadamente unos 20 minutos antes para permitir que alcancen la temperatura ambiente (23°C-27°C). Antes de la dilución, se inspeccionará cada vial para detectar partículas o cambios de color. La solución transparente a ligera opalescente o incolora a amarillo pálido debe estar libre de partículas visibles. No utilizar el producto si el líquido en la ampolleta está turbio o tiene partículas. Se extraerá y desechará un volumen de la solución de cloruro sódico al 0,9% para infusión de una bolsa de infusión de 250 ml igual al volumen total de NAGLAZYME® que se añade. Se considerará el uso de bolsas de infusión de 100ml en pacientes sensibles a la sobre carga de volumen de líquido y en aquellos con un peso corporal inferior a 20 kg; en dichos casos, se reducirá la velocidad de infusión (ml/min) demodo que la duración total no sea menor de 4 horas. Si se usan bolsas de 100 ml, el volumen de NAGLAZYME® puede añadirse directamente a la bolsa de infusión. El volumen de NAGLAZYME® se añadirá lentamente a la solución para infusión de cloruro sódico 9 mg/ml (0,9%). La solución se mezclará suavemente antes de la infusión. Se inspeccionará visualmente la solución para detectar partículas antes del uso. Sólo se usarán las soluciones transparentes e incoloras sin partículas visibles.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: No se han notificado casos de sobredosis de NAGLAZYME®. Varios pacientes han recibido la dosis total de NAGLAZYME® aproximadamente al doble de la velocidad de infusión recomendada, sin reacciones adversas aparentes.
Presentación(es): Caja de cartón con un frasco ámpula.
Recomendaciones sobre almacenamiento: Conservar en refrigeración (entre 2°C -8°C). No congelar ni agitar el frasco ámpula. Solución diluida: NAGLAZYME® debe usarse inmediatamente. Si no se usa inmediatamente, los tiempos y condiciones de conservación en uso son responsabilidad del usuario. No deben exceder de 24 horas a 2°C-8°C, seguidas de hasta 24 horas a temperatura ambiente, no más de 27°C durante la administración.
Leyendas de protección: Dosis: la que el médico señale. Vía de administración: intravenosa, por infusión. Léase instructivo anexo. No se deje al alcance de los niños. Su venta requiere receta médica. No se use en el embarazo. No se administre si la solución no es transparente o tiene partículas en suspensión o sedimento. No se administre si el cierre ha sido violado. Una vez diluido, si no se administra todo el producto, deseche el sobrante. Consérvese en refrigeración de 2°C-8°C. No congelar ni agitar el frasco ámpula.
Nombre y domicilio del laboratorio: Hecho en los Estados Unidos por: Biomarin Pharmaceutical Inc. 46 Galli Drive, Novato, CA 94949, USA. Importado y distribuido en México por: Específicos Stendhal S.A. de C.V. Escorpión Lote 10, Fracc. Ind. San Isidro, Chicoloapan, Edo. de México 56370.
Número de registro del medicamento: 002HU M2009 SSA IV.
Clave de IPPA: No. de soicitud 093300405B0001