NEXAVAR®
BAYER HEALTH C.
Denominación genérica: Sorafenib.
Forma farmacéutica y formulación: Cada comprimido contiene: tosilato de sorafenib equivalente a 200 mg de sorafenib. Excipiente c.b.p. 1 comprimido.
Indicaciones terapéuticas: NEXAVAR® está indicado en el tratamiento de pacientes con carcinoma celular renal avanzado.
Farmacocinética y farmacodinamia: Farmacocinética: después de la administración de NEXAVAR® comprimidos 200 mg, la biodisponibilidad relativa promedio es de 38-49%, en comparación con una solución oral de sorafenib. La vida media de eliminación de sorafenib es de 25-48 horas aproximadamente. La dosificación múltiple de sorafenib durante 7 días da lugar a una acumulación de 2,5 a 7 veces en comparación con la administración de una dosis única. Las concentraciones plasmáticas de sorafenib en estado de equilibrio se alcanzan en 7 días, con una relación de máximo mínimo de las concentraciones promedio inferior a 2. Absorción y distribución: después de la administración oral, sorafenib alcanza niveles plasmáticos máximos en 3 horas, aproximadamente. Cuando se administra con una dieta moderada en grasas, la biodisponibilidad es similar a la registrada en ayunas. Con una dieta rica en grasas, la biodisponibilidad de sorafenib se reduce en 29% en comparación con la administración en ayunas. La Cmáx y ABC promedio aumentan por debajo de la proporcionalidad con dosis superiores a 400 mg administrados dos veces al día por vía oral. La unión a proteínas plasmáticas humanas in vitro es 99,5%. Metabolismo y eliminación: NEXAVAR® se metaboliza principalmente en el hígado por metabolismo oxidativo mediado por CYP3A4, así como glucuronización mediada por UGT1A9. Sorafenib supone alrededor del 70-85% de los analitos circulantes en plasma en estado de equilibrio. Se han identificado 8 metabolitos de sorafenib, de los cuales cinco se han determinado en plasma. El principal metabolito circulante de sorafenib en plasma, N-óxido de piridina, demuestra una potencia in vitro similar a la del sorafenib y supone 9-16% aproximadamente de los analitos circulantes en estado de equilibrio. Después de la administración oral de una dosis de 100 mg de una formulación en solución de sorafenib, 96% de la dosis se recuperó en 14 días, el 77% de la dosis eliminado por heces y el 19% por orina como metabolitos glucuronizados. Sorafenib excretado como forma inalterada corresponde a 51% de la dosis y se recuperó en heces, pero no en orina. Estudios in vitro en la inhibición enzimática: los estudios realizados en microsomas hepáticos humanos demostraron que sorafenib es un inhibidor competitivo de CYP2C19, CYP2D6 y CYP3A4. Sorafenib puede incrementar los niveles plasmáticos de fármacos que son sustratos de estas enzimas. Sorafenib inhibe la glucuronización a través de las vías UGT1A1 y UGT1A9. La exposición sistémica a sustratos de UGT1A1 y UGT1A9 puede verse incrementada cuando se administran concomitantemente con sorafenib. Sorafenib inhibe CYP2B6 y CYP2C8 in vitro con valores Ki de 6 y 1-2 mM, respectivamente. La exposición sistémica a sustratos de CYP2B6 y CYP2C8 puede verse incrementada cuando se coadministran con sorafenib. Sustratos CYP2C9: los estudios realizados con microsomas hepáticos humanos han demostrado que sorafenib es un inhibidor competitivo de CYP2C9 con un valor Ki de 7-8 mM. El posible efecto de sorafenib sobre un sustrato CYP2C9 se valoró en pacientes a los que se les administró sorafenib o placebo en combinación con warfarina. Los cambios promedios a partir de los valores basales del INR-TP no fueron superiores en los pacientes con sorafenib en comparación con los pacientes con placebo, lo cual sugiere que sorafenib puede no ser un inhibidor in vivo de CYP2C9. Inhibidores de CYP3A4: ketoconazol (400 mg), un potente inhibidor de CYP3A4, administrado una vez al día durante 7 días a voluntarios varones sanos no alteró el ABC promedio de una dosis única de 50 mg de sorafenib. Estudios in vitro de la inducción de enzimas CYP: las actividades CYP1A2 y CYP3A4 no se vieron alteradas tras el tratamiento de hepatocitos humanos cultivados con sorafenib, indicando que es poco probable que sorafenib sea un inductor de CYP1A2 y CYP3A4. Farmacocinética en poblaciones especiales: adultores mayores (mayores de 65 años) y sexo: el análisis de los datos demográficos sugieren que no es necesario efectuar ajustes de dosis por edad o sexo. Pacientes pediátricos: no se dispone de datos farmacocinéticos en pacientes pediátricos. Insuficiencia hepática: sorafenib se elimina principalmente a través del hígado. En pacientes con insuficiencia hepática leve (Child-Pugh A, N=14) o moderada (Child-Pugh B, N=8), los valores de exposición se situaron dentro del rango observado en pacientes sin insuficiencia hepática. La farmacocinética de Sorafenib no se ha estudiado en pacientes con insuficiencia hepática grave (Child-Pugh C) Insuficiencia renal: en pacientes con función renal normal (N=71) y en pacientes con insuficiencia renal leve (CrCl > 50-80 ml/min, N=24) o moderada (CrCl 30-50 ml/min, N=4), no se observó ninguna relación entre el ABC de sorafenib en estado de equilibrio y la función renal a dosis de 400 mg dos veces al día. La farmacocinética de sorafenib no se ha estudiado en pacientes con insuficiencia renal grave (CrCl < 30 ml/min) o en pacientes sometidos a diálisis (Ver Precauciones Generales). Farmacodinamia: mecanismo de acción: sorafenib es un inhibidor multicinasa que reduce la proliferación celular tumoral in vitro. Sorafenib inhibe el crecimiento tumoral del carcinoma de células renales murínicas, RENCA y un amplio espectro de xenoinjertos tumorales humanos en ratones atímicos acompañado de una reducción de la angiogénesis tumoral. Sorafenib inhibe la actividad de las dianas presentes en la célula tumoral (CRAF, BRAF, V600E BRAF, KIT y FLT-3) y en la vasculatura tumoral (CRAF, VEGFR-2, VEGFR-3 y PDGFR-ß). Las cinasas RAF son cinasas serina/treonina, mientras que KIT, FLT-3, VEGFR-2, VEGFR-3 y PDGFR-ß son receptores tirosina cinasa. La mutación de BRAF ha sido asociada con melanoma, KIT se ha asociado a tumores del estroma gastrointestinal y FLT-3 se ha asociado a la leucemia mielógena aguda. Eficacia clínica: la seguridad y eficacia de NEXAVAR® en el tratamiento de carcinoma celular renal avanzado (RCC) fueron analizadas en dos estudios clínicos controlados aleatorizados. El estudio 1 fue un estudio de Fase III, multicéntrico, internacional, aleatorizado, doble ciego, controlado con placebo en 903 pacientes. Los parámetros primarios incluyeron supervivencia global (OS) y supervivencia sin progresión (PFS) El índice de respuesta del tumor fue un parámetro secundario. Los pacientes fueron aleatorizados a NEXAVAR® 400 mg dos veces al día (N=451) o placebo (N=452). En un análisis intermedio de los datos de supervivencia, basado en 220 muertes, se encontró una mejoría estimada del 39% en la supervivencia global en pacientes que recibieron NEXAVAR® contra placebo. La proporción de riesgo estimada (riesgo de muerte con NEXAVAR® comparado con placebo) fue 0,72 (IC95%, 0,55-0,95; p=0,018) El umbral de significancia estadística fue p < 0,0005. La supervivencia sin progresión incluyó 769 pacientes aleatorizados a NEXAVAR® 400 mg dos veces al día (N=384) o placebo (N=385). La supervivencia sin progresión fue dos veces mayor para pacientes aleatorizados a NEXAVAR® (167 días) comparado con placebo (84 días) El índice estimado de riesgo (riesgo de progresión con NEXAVAR® comparado a placebo) fue 0,44 (IC95%: 0,35- 0,55 p < 0,000001). El efecto de NEXAVAR® en la supervivencia sin progresión fue consistente con el subconjunto, incluyendo pacientes sin terapia precia de IL-2 o interferón (N=137), 65 pacientes que recibieron NEXAVAR® y 72 pacientes tratados con placebo) para los cuales la supervivencia sin progresión promedio fue 172 días para NEXAVAR® comparado con 85 días para placebo. El estudio 2 fue un estudio Fase II aleatorizado, de descontinuación, en pacientes con tumores metastáticos, incluyendo carcinoma celular renal. El parámetro primario fue el porcentaje de pacientes aleatorizados (N= 65) que permanecieron sin progresión a las 24 semanas. La supervivencia sin progresión fue mayor en pacientes aleatorizados a NEXAVAR® (163 días) que en pacientes aleatorizados a placebo (41 días) (p=0,0001, indice de riesgo= 0,29) La tasa de supervivencia sin progresión fue significativamente mayor en el grupo de NEXAVAR® (50%) que en el grupo placebo (18%) (p=0,0077).
Contraindicaciones: NEXAVAR® está contraindicado en pacientes con hipersensibilidad severa conocida o sospechada a sorafenib o cualquiera de los componentes de la formulación.
Precauciones generales: Embarazo: durante el tratamiento con NEXAVAR®, las mujeres en edad fértil deberán evitar embarazarse. Asimismo, el médico deberá advertir a las mujeres embarazadas sobre los posibles riesgos para el feto, entre los que se incluyen malformaciones graves (teratogenicidad), falta de crecimiento y muerte fetal (embriotoxicidad). NEXAVAR® no debe ser administrado durante el embarazo. El médico sólo debe considerar su uso cuando los beneficios potenciales justifiquen el riesgo para el feto (Ver Precauciones o restricciones de uso durante el embarazo y la lactancia). Con base a los mecanismos propuestos de inhibición multicinasas y a los múltiples eventos adversos observados en animales a niveles de exposición significativamente por debajo de la dosis diaria, se deberá asumir que NEXAVAR® puede causar daño al feto cuando es administrado a mujeres embarazadas. Durante el tratamiento con sorafenib, se deberá suspender la lactancia. Toxicidad dermatológica: las reacciones adversas más comunes son las reacciones cutáneas en manos-pies (eritrodisestesia palmo-plantar) y exantema. Estas reacciones suelen ser de grado 1 y 2 de acuerdo a los criterios comunes de toxicidad (CTC, Common Toxicity Criteria) del Instituto Nacional del Cáncer (National Cancer Institute) y aparecen durante las primeras seis semanas durante el tratamiento con NEXAVAR®. El control de la toxicidad dermatológica puede incluir el tratamiento tópico para el alivio sintomático y/o interrupción temporal del tratamiento de la dosis de sorafenib o en casos graves o persistentes, la interrupción permanente del tratamiento. Hipertensión: se observó un incremento en la incidencia de hipertensión en pacientes tratados con sorafenib. En general, la hipertensión fue de leve a moderada, se produjo al inicio del tratamiento y fue controlable con un tratamiento antihipertensivo estándar. La presión sanguínea deberá monitorearse regularmente y ser tratada, en caso necesario, según las prácticas médicas estándar. En caso de hipertensión grave o persistente o de crisis hipertensivas a pesar del tratamiento antihipertensivo adecuado, se deberá considerar la interrupción permanente del tratamiento con NEXAVAR®. Hemorragia: puede ocurrir un incremento del riesgo de hemorragias después de la administración de sorafenib. La incidencia de acontecimientos hemorrágicos graves es poco común. Si un acontecimiento hemorrágico requiere intervención médica, es recomendable considerar la interrupción permanente del tratamiento con NEXAVAR®. Warfarina: en algunos pacientes administrados concomitante con warfarina durante el tratamiento con sorafenib, se han descrito acontecimientos hemorrágicos poco frecuentes o incrementos en el Indice Internacional Normalizado (INR, International Normalized Ratio). En pacientes administrados concomitantemente con warfarina, se deberá monitorear regularmente los cambios en el tiempo de protombina, INR y episodios hemorrágicos clínicos. Complicaciones en la curación de heridas: no se han realizado estudios formales sobre el efecto de sorafenib en la curación de heridas. En pacientes sometidos a intervenciones quirúrgicas mayores, se recomienda la interrupción temporal del tratamiento con sorafenib como medida de precaución. La experiencia clínica con relación al intervalo de tiempo para reiniciar el tratamiento tras una intervención quirúrgica mayor es limitada. Por ello, la decisión de reiniciar el tratamiento con sorafenib después de una intervención quirúrgica mayor debe basarse en el dictamen clínico de la curación adecuada de la herida. Isquemia y/o infarto al miocardio: en el estudio 1, se observó un incremento en la incidencia de isquemia/infarto al miocardio de tratamiento emergente en pacientes tratados con NEXAVAR® (2,9%) en comparación con un grupo placebo (0,4%). Los pacientes que presentaron enfermedad coronaria inestable o infarto al miocardio reciente fueron excluidos de este estudio. Se deberá considerar la suspensión temporal o permanente del tratamiento con NEXAVAR® en pacientes que hayan desarrollado isquemia y/o infarto al miocardio. Alteración hepática: no existen datos de alteración hepática severa (Chile Pugh C). Debido a que NEXAVAR® es eliminado principalmente por vía hepática, la exposición puede incrementar en pacientes con alteración hepática severa. Interacciones medicamentosas: se recomienda precaución en la administración de sorafenib conjuntamente con compuestos que se metabolizan/eliminan principalmente a través de la vía UGT1A1 (p. ej., Irinotecan) (ver Interacciones Medicamentosas y de otro género).
Restricciones de uso durante el embarazo y la lactancia: Embarazo: no se han realizado estudios adecuados y bien controlados en mujeres embarazadas administradas con sorafenib. Los estudios en animales han demostrado toxicidad en la reproducción, incluyendo malformaciones. Estudios en ratas demostraron que sorafenib y sus metabolitos cruzan la placenta y se anticipa que sorafenib inhiba la angiogénesis en el feto. Con base a los mecanismos propuestos de inhibición multicinasas y a los múltiples efectos adversos observados en animales a niveles de exposición significativamente por debajo de la dosis clínica, se deberá asumir que NEXAVAR® causa daño fetal cuando se administra a mujeres embarazadas. Durante el tratamiento con NEXAVAR®, las mujeres en edad fértil deberán evitar embarazarse. Asimismo, el médico deberá advertir a las mujeres sobre los posibles riesgos para el feto, entre los que se incluyen malformaciones graves (teratogenicidad), falta de crecimiento y muerte fetal (embriotoxicidad). NEXAVAR® no debe ser usado durante el embarazo. El médico sólo debe considerar su uso cuando los beneficios potenciales justifiquen el riesgo para el feto (ver Precauciones generales). Mujeres en edad fértil: se ha demostrado que sorafenib es teratogénico y embriotóxico en animales. Se deberá asegurar la adecuada contracepción durante el tratamiento con NEXAVAR® y, por lo menos, 2 semanas después de completar el tratamiento. Lactancia: se desconoce si sorafenib es excretado en la leche humana. En animales, sorafenib y/o sus metabolitos fueron excretados en la leche materna. Considerando que varios fármacos son excretados en la leche materna y debido a que los efectos de sorafenib en los lactantes no han sido estudiados, se deberá discontinuar la lactancia materna durante el tratamiento con sorafenib. Fertilidad: los resultados de estudios animales indican que sorafenib puede alterar la fertilidad masculina y femenina. Efecto en la capacidad para operar maquinaria: no se han realizado estudios relacionados con el efecto de NEXAVAR® en la capacidad para manejar u operar maquinaria. No hay evidencia que sorafenib afecte la capacidad para manejar u operar maquinaria.
Reacciones secundarias y adversas: El perfil de seguridad global de sorafenib está basado en 1.286 pacientes con cáncer que recibieron NEXAVAR® como agente único y 165 pacientes que recibieron NEXAVAR® concomitantemente con quimioterapia. La tabla 1 incluye el porcentaje de pacientes que experimentaron reacciones adversas de tratamiento emergente que se reportaron en al menos el 10% de los pacientes que recibieron NEXAVAR® en el estudio 1 (véase Apartado Propiedades Farmacodinámicas-Eficacia Clínica). Un total de 346 pacientes fueron tratados con NEXAVAR® como monoterapia por más de 6 meses. Un total de 664 pacientes con carcinoma celular renal avanzado recibieron sorafenib como monoterapia de los cuales, 215 fueron tratados por al menos 6 meses. Las reacciones adversas de tratamiento emergente de Grado 3 de CCTAA fueron reportados en 31% de los pacientes que recibieron NEXAVAR® comparado a 22% de los pacientes que recibieron placebo. Las reacciones de tratamiento emergente de Grado 4 fueron reportados en 7% de los pacientes que recibieron NEXAVAR® comparado a 6% de los pacientes que recibieron placebo.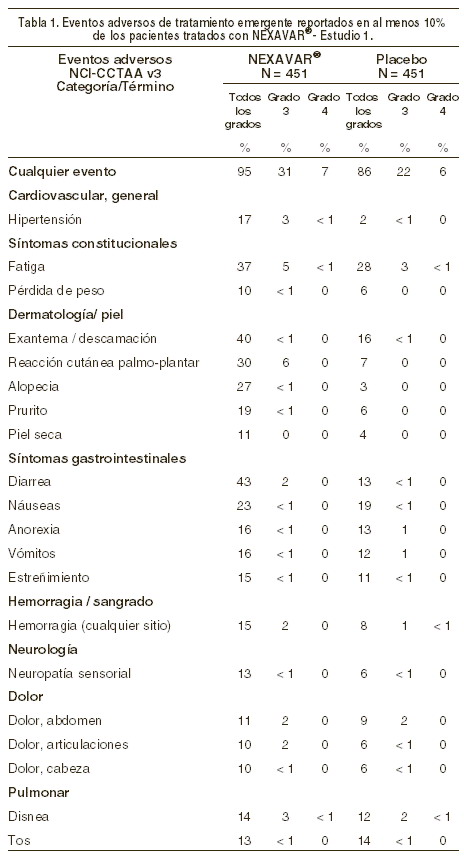
El índice de eventos adversos (incluyendo eventos asociados con enfermedad progresiva) que resultaron en la discontinuación permanente fue similar en los grupos de NEXAVAR® y placebo (10% de los pacientes tratados con NEXAVAR® y 8% en pacientes tratados con placebo). La seguridad fue verificada con los datos en el estudio en Fase II que incluyó 638 pacientes tratados con NEXAVAR®, que incluyó 202 pacientes con carcinoma celular renal avanzado, 137 pacientes con carcinoma hepatocelular y 299 pacientes con otro tipo de cáncer. Los eventos adversos más comunes relacionados con el fármaco reportado en pacientes tratados con NEXAVAR® en estos datos fueron rash (38%), diarrea (37%), reacciones palmo-plantar (35%) y fatiga (33%). Los índices respectivos de eventos relacionados con el fármaco de CTC (v.20) de Grado 3 y 4 en pacientes tratados con NEXAVAR® fue 37% y 3% respectivamente. Datos adicionales procedentes de múltiples estudios clínicos: se observaron los siguientes eventos adversos relacionados con el fármaco y alteraciones a las pruebas de laboratorio registrados en los estudios clínicos realizados en 1286 pacientes que recibieron sorafenib como monoterapia (muy frecuente: ≥10%; frecuente: 1% a < 10%; poco frecuente: 0,1% a < 1%). Cardiovascular: poco frecuente: crisis hipertensiva, isquemia y/o infarto al miocardio. Dermatológicas: muy frecuente: eritema. Frecuente: dermatitis exfoliativa, acné, enrojecimiento. Poco frecuente: foliculitis, eczema y eritema multiforme. Digestivo: muy frecuente: incremento de lipasa, incremento de amilasa. Frecuente: mucositis, estomatitis (incluyendo resequedad en la boca y glosodinia), dispepsia, disfagia. Poco frecuente: pancreatitis, reflujo gastrointestinal, gastritis. La elevación de lipasa es muy frecuente (41%); un diagnóstico de pancreatitis no se deberá basar únicamente en resultados anormales a las pruebas de laboratorio. Alteraciones generales: muy frecuente: astenia, dolor (incluyendo dolor en la boca, huesos y muscular). Frecuente: disminución del apetito, síntomas similares a la influenza, pirexia. Poco frecuente: infección. Hematología: muy frecuente: leucopenia, linfopenia. Frecuente: anemia, neutropenia, trombocitopenia. Poco frecuente: INR anormal. Hipersensibilidad: poco frecuente: reacciones de hipersensibilidad (incluyendo reacciones cutáneas y urticaria). Metabólico y nutricional: muy frecuente: hipofosfatemia. Frecuente: incremento transitorio de transaminasas. Poco frecuente: deshidratación, hiponatremia, incremento transitorio en fosfatasa alcalina, incremento de bilirrubina (incluyendo ictericia), hipotiroidismo. Músculo-esquelético: frecuente: artralgia, mialgia. Sistema nervioso y psiquiátricas: frecuente: depresión. Poco frecuente: tinnitus. Reproducción: frecuente: disfunción eréctil. Poco frecuente: ginecomastia. Respiratorio: frecuente: ronquera. Poco frecuente: rinorrea. Adicionalmente, los siguientes eventos adversos de significancia clínica fueron reportados infrecuentemente durante los estudios clínicos con NEXAVAR®: hemorragia cerebral, ataque isquémico reciente, falla cardíaca, arritmia, tromboembolismo y falla renal aguda. Para estos eventos, la relación causal con NEXAVAR® no ha sido establecida.
Interacciones medicamentosas y de otro género: Inductores del citocromo CYP3A4: no se dispone de información clínica del efecto de inductores del citocromo CYP3A4 en la farmacocinética de sorafenib. La actividad de los inductores del CYP3A4 (p. e. rifampicina, Hypericum perforatum o hierba de San Juan, fenitoína, carbamazepina, fenobarbital y dexametasona) puede incrementar el metabolismo de sorafenib y, por lo tanto, disminuir la concentración de sorafenib. Inhibidores del citocromo CYP3A4: ketoconazol, un potente inhibidor del citocromo CYP3A4, administrado una vez al día durante 7 días a voluntarios varones sanos, no interfiere con el ABC promedio de una dosis única de 50 mg de sorafenib. Las interacciones de sorafenib con inhibidores del citocromo CYP3A4 son poco probables. Sustratos de CYP2C9: se evaluó el posible efecto de sorafenib sobre la warfarina, un sustrato de CYP2C9, en pacientes tratados con sorafenib, en comparación con pacientes tratados con placebo. El tratamiento concomitante con sorafenib y warfarina no dio lugar a cambios en el INR-TP promedio con placebo. Sin embargo, es necesario controlar regularmente el INR en pacientes administrados con warfarina. Sustratos selectivos de isoformas CYP: la administración concomitante de midazolam, dextrometorfano y omeprazol (sustratos de los citocromos CYP3A4, CYP2D6 y CYP2C19, respectivamente) después de la administración de sorafenib durante 4 semanas, no alteró la exposición de estos agentes. Esto indica que sorafenib no es inhibidor o inductor de estas isoenzimas del citocromo P-450. Combinación con otros agentes anti-neoplásicos: en diversos estudios clínicos, sorafenib ha sido administrado concomitantemente con diferentes agentes antineoplásicos (gemcitabina, oxaliplatino, doxorubicina e irinotecan) a los regímenes de dosis habituales. Sorafenib no tiene efecto en la farmacocinética de gemcitabina u oxaliplatino. El tratamiento concomitante con sorafenib incrementó en 21% el ABC de doxorubicina. Cuando se administró concomitantemente con irinotecan, cuyo metabolito activo SN-38 es metabolizado vía UGT1A1, se produjo incremento de 67-120% el ABC de SN-38 e incremento de 26-42% en el ABC de irinotecan. La significancia clínica de estos hallazgos es desconocida.
Alteraciones en los resultados de pruebas de laboratorio: Con mucha frecuencia, se han descrito aumentos de los niveles de lipasas y amilasas. En el estudio 1, se produjeron aumentos de lipasas, CCTAA de grado 3 o 4 en el 12% de los pacientes en el grupo de sorafenib, en comparación con el 7% de los pacientes en el grupo de placebo. Se refirieron aumentos de la amilasa CCTAA de grado 3 o 4 en el 1% de los pacientes del grupo con sorafenib, en comparación con el 3% de los pacientes en el grupo placebo. En 2 de los 451 pacientes tratados con sorafenib se describió una pancreatitis clínica (CCTAA de grado 4) y en 1 de los 451 pacientes (CCTAA de grado 2) en el grupo con placebo del estudio 1. La hipofosfatemia fue un resultado frecuente de laboratorio, que se pudo observar en el 45% de los pacientes tratados con sorafenib, en comparación con 11% de los pacientes con placebo. En 13% de los pacientes tratados con sorafenib y en 3% de los pacientes del grupo con placebo se produjo una hipofosfatemia CCTAA de grado 3 (1-2 mg/dl). No se dieron casos de hipofosfatemia CCTAA de grado 4 ( < 1 mg/dl) en ningún paciente de los grupos con sorafenib o con placebo. Se desconoce la etiología de la hipofosfatemia asociada a sorafenib. Se registró linfopenia CCTAA de grado 3 o 4 en 13% de los pacientes tratados con sorafenib y 7% en pacientes tratados con placebo, neutropenia en el 5% de los pacientes tratados con sorafenib y en el 2% de los pacientes con placebo, anemia en el 2% de los pacientes tratados con sorafenib y en el 4% de los pacientes con placebo y trombocitopenia en el 1% de los pacientes tratados con sorafenib y en el 0% de los pacientes con placebo.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: El perfil de seguridad preclínica de sorafenib se evaluó en ratones, ratas, perros y conejos. Los estudios de la toxicidad de dosis repetida mostraron cambios leves a moderados (degeneraciones y regeneraciones) en diferentes órganos. Tras la administración de dosis repetidas a perros jóvenes y en crecimiento, se observaron efectos sobre huesos y dientes. Los cambios consistieron en un engrosamiento irregular de la placa de crecimiento femoral a una dosis diaria de sorafenib de 600 mg/m2 de área de superficie corporal (equivalente a 1,2 veces la dosis clínica recomendada de 500 mg/m2 sobre la base del área de superficie corporal), hipocelularidad de la médula ósea adyacente a la placa de crecimiento alterada a dosis de 200 mg/m2/día y alteraciones en la composición de la dentina a dosis de 600 mg/m2/día. En perros adultos no se indujeron efectos similares. Se obtuvieron efectos genotóxicos positivos con sorafenib en un ensayo en células mamíferas in vitro (ovario de hámster chino) en cuanto a la clastogenicidad (aberraciones cromosomales) en presencia de activación metabólica. Un producto intermedio del proceso de fabricación, que también se encuentra en el medicamento final ( < 0,15%), dio positivo en cuanto a mutagénesis en un ensayo de células bacterianas in vitro (prueba de Ames). Sorafenib no fue genotóxico en la prueba de Ames (el material contenía el intermedio en un 0,34%) ni tampoco en el ensayo del micronúcleo murínico in vivo. No se han realizado estudios de carcinogenicidad con sorafenib. No se han realizado estudios específicos con sorafenib en animales para evaluar el efecto sobre la fertilidad. Sin embargo, cabe esperar un efecto adverso sobre la fertilidad masculina y femenina porque los estudios de dosis repetidas en animales han demostrado cambios en los órganos reproductores masculinos y femeninos. Los cambios típicos consistieron en signos de degeneración y retardo en testículos, epidídimos, próstata y vesículas seminales de las ratas con efectos evidentes a una dosis diaria de sorafenib de 150 mg/m2 de área de superficie corporal (equivalente a aproximadamente 0,3 veces la dosis clínica recomendada de 500 mg/m2 sobre una base del área de superficie corporal). Las ratas hembra mostraron una necrosis central del cuerpo lúteo e interrupción del desarrollo folicular de los ovarios con un efecto mínimo observado a dosis de 30 mg/m2/día. Los perros mostraron degeneración tubular en los testículos a dosis de 600 mg/m2/día y oligospermia a 1.200 mg/m2/día. Sorafenib ha demostrado ser embriotóxico y teratogénico cuando se administra a ratas y conejos. Los efectos observados incluyen reducción de los pesos corporales maternos y fetales, un aumento del número de reabsorciones fetales y un aumento del número de malformaciones externas y viscerales. Los resultados adversos fetales se observaron a una dosis oral de 6 mg/m2/día en ratas y 36 mg/m2/día en conejos.
Dosis y vía de administración: La dosis diaria recomendada es 800 mg repartidos en dos tomas de 400 mg cada una (2 comprimidos de NEXAVAR®) cada 12 horas. NEXAVAR® puede ser ingerido sin alimentos o con alimentos (con un contenido moderado en grasa). Vía de administración: oral. Ingerir los comprimidos enteros con un vaso de agua (250 ml). Duración del tratamiento: el tratamiento con NEXAVAR® deberá continuarse hasta que el paciente no sea beneficiado clínicamente con la terapia o hasta que se manifieste una toxicidad no aceptable. Titulación de dosis, ajuste de dosis y recomendaciones especiales durante el monitoreo: el control en caso de sospecha de reacciones adversas del fármaco puede requerir la interrupción temporal y/o reducción de la dosis durante el tratamiento con NEXAVAR®. Cuando la reducción de dosis sea necesaria, la dosis de NEXAVAR® puede ser reducida a 400 mg al día (2 comprimidos de 200 mg) (ver Precauciones generales). Modificación de la dosis sugerida en caso de toxicidad cutánea.
Niños: la eficacia y seguridad de NEXAVAR® en pacientes pediátricos no han sido establecidas. Adultos mayores (mayores a 65 años), sexo y peso corporal: no se requiere ajuste de dosis con base a la edad, sexo o peso corporal del paciente. Insuficiencia hepática: no se requiere ajuste de dosis en pacientes con alteración hepática leve Child-Pugh A y B. No se ha estudiado el uso de sorafenib en pacientes con alteración hepática severa Child-Pugh C. Alteración renal: no se requiere ajuste dosis en pacientes con alteración renal leve a moderada. El uso de sorafenib no ha sido estudiado en pacientes con insuficiencia renal severa o en pacientes sometidos a diálisis.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: No existe ningún tratamiento específico para la sobredosis con NEXAVAR®. La dosis máxima de sorafenib estudiada clínicamente es de 800 mg, dos veces al día. Las reacciones adversas observadas a esta dosis fueron principalmente diarrea y eventos dermatológicos. En el caso de sospecha de sobredosis, debe interrumpirse la administración de sorafenib e instaurarse un tratamiento de soporte.
Presentación(es): Caja de cartón con 28, 56 y 112 comprimidos de 200 mg en envase de burbuja de polipropileno (blíster). Caja de cartón con 28, 56 y 112 comprimidos de 200 mg en envase de burbuja (blíster).
Recomendaciones sobre almacenamiento: Consérvese a temperatura ambiente a no más de 25° C y en lugar fresco y seco.
Leyendas de protección: Literatura exclusiva para médicos. Manténgase fuera del alcance de los niños. Su venta requiere receta médica. No se administre durante el embarazo y la lactancia. Consérvese a temperatura ambiente a no más de 25°C y en lugar fresco y seco.
Nombre y domicilio del laboratorio: Hecho en Alemania por: Bayer Healthcare AG. 51368 Leverkusen, Alemania. Distribuido por: Bayer de México, S.A. de C.V. Miguel de Cervantes Saavedra No. 259, C.P. 11520, México, D.F.
Número de registro del medicamento: 082M2006 SSA IV.
Clave de IPPA: CEAR-06330060100164/R2006