NUBRENZA
UCB
Denominación genérica: Rotigotina
Forma farmacéutica y formulación: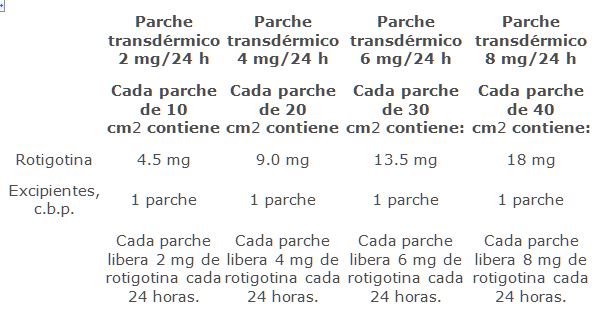
Indicaciones terapéuticas: NUBRENZA® está indicado para: El tratamiento de los signos y síntomas de la enfermedad de Parkinson idiopática.
Farmacocinética y farmacodinamia: Farmacocinética: Absorción: Después de la aplicación se libera rotigotina continuamente desde el parche transdérmico y se absorbe a través de la piel. Las concentraciones en equilibrio se alcanzan después de uno o dos días de aplicación del parche y se mantienen estables mediante la aplicación una vez al día cuando se lleva puesto el parche durante 24 horas. La concentración plasmática de rotigotina aumenta proporcionalmente a la dosis en un intervalo de 1 mg/24 h a 24 mg/24 h. Aproximadamente el 45% del principio activo contenido en el parche se libera en la piel en 24 horas. La biodisponibilidad absoluta después de la aplicación transdérmica es del 37%. La rotación del lugar de aplicación del parche puede provocar diferencias diarias en las concentraciones plasmáticas. Las diferencias de la biodisponibilidad de rotigotina variaron del 1% (cadera frente a abdomen) al 46% (hombro frente a muslo). No obstante, no hay indicios de un impacto relevante en la evolución clínica. Distribución: La unión in vitro de rotigotina a las proteínas plasmáticas es aproximadamente del 92%. El volumen aparente de distribución en el ser humano es de aproximadamente 84 L/kg. Metabolismo: Rotigotina se metaboliza en un alto porcentaje. Rotigotina se metaboliza mediante N-desalquilación y también mediante conjugación directa y secundaria. Los resultados obtenidos in vitro indican que hay varias isoformas de las enzimas CYP capaces de catalizar la N-desalquilación de rotigotina. Los metabolitos principales son conjugados sulfato y glucurónido del compuesto original y metabolitos N-desalquilados, que son biológicamente inactivos. Eliminación: El 71% de la dosis de rotigotina se excreta por la orina y una cantidad menor, en torno al 23%, se excreta por las heces. La depuración de rotigotina después de la administración transdérmica es de 10 L/min y su vida media de eliminación total es de 5 a 7 horas. El perfil farmacocinético muestra una eliminación bifásica con una vida media inicial de aproximadamente 2 a 3 horas. Como el parche se administra por vía transdérmica, no se esperan efectos debido a las comidas o la situación gastrointestinal. Poblaciones especiales Como el tratamiento con NUBRENZA® comienza con una dosis baja y se ajusta gradualmente según la tolerabilidad clínica hasta obtener un efecto terapéutico óptimo, no es necesario ajustar la dosis según el sexo, el peso o la edad. Insuficiencia renal: En pacientes con insuficiencia renal de leve a severa, no se observaron incrementos relevantes en las concentraciones plasmáticas de rotigotina. Las concentraciones plasmáticas de los conjugados de rotigotina y sus metabolitos de alquilo, aumentan a medida que se deteriora la función renal. No obstante, es improbable que estos metabolitos contribuyan con los efectos clínicos. Insuficiencia hepática: En pacientes con insuficiencia hepática moderada, no se observaron aumentos relevantes de los niveles plasmáticos de la rotigotina. No se investigó el efecto de NUBRENZA® en pacientes con insuficiencia hepática severa. Farmacodinamia: Grupo farmacoterapéutico: Agonistas de dopamina, rotigotina; código ATC: N04BC09. Rotigotina es un agonista de dopamina no ergotamínico para el tratamiento de los signos y síntomas de la enfermedad de Parkinson. Mecanismo de acción: Se cree que la rotigotina promueve su efecto benéfico en la enfermedad de Parkinson mediante la activación de los receptores D3, D2 y D1 del caudado-putamen en el cerebro. Farmacodinamia: Con respecto a la actividad funcional y los diversos subtipos de receptor y su distribución en el cerebro, la rotigotina se describe como agonista de los receptores D2 y D3 actuando también en los receptores D1, D4 y D5. Entre los receptores no-dopaminérgicos, la rotigotina mostró antagonismo en los receptores alfa 2B y agonismo en los receptores 5HT1A, lo cual contribuiría también a su perfil de eficacia in vivo. No hay actividad de la rotigotina en el receptor 5HT2B.
Contraindicaciones: Hipersensibilidad a la sustancia activa o a cualquiera de los excipientes.
Precauciones generales: Imagen por resonancia magnética y cardioversión. La capa externa de NUBRENZA® contiene aluminio. Para evitar quemaduras en la piel se debe retirar el parche de NUBRENZA® cuando el paciente se someta a un estudio de imagen por resonancia magnética (RM) o cardioversión. Hipotensión ortostática: Los agonistas de la dopamina alteran la regulación sistémica de la presión arterial, por lo que pueden provocar hipotensión postural u ortostática. Estos episodios también se han observado durante el tratamiento con NUBRENZA®, pero la incidencia fue similar a la observada en los pacientes tratados con placebo. Se debe considerar el monitoreo de la presión sanguínea. Síncope: En los estudios clínicos se ha observado síncopes asociados con NUBRENZA®, aunque con una tasa similar a la de los pacientes tratados con placebo. Inicio súbito del sueño y somnolencia: El tratamiento con NUBRENZA® se ha asociado a somnolencia y episodios de inicio repentino del sueño. Se ha reportado que el inicio repentino del sueño puede aparecer durante las actividades cotidianas, a veces sin signos previos de aviso. El médico responsable debe reevaluar continuamente la aparición de somnolencia o adormecimiento, ya que los pacientes no reconocen su presencia hasta que se les interroga directamente. Trastornos en el control de los impulsos y otros relacionados: Los pacientes deben ser monitoreados regularmente por la aparición de trastornos compulsivos. Los pacientes y sus cuidadores deben tener en cuenta que, en los pacientes tratados con agonistas dopaminérgicos para la enfermedad de Parkinson, incluyendo NUBRENZA® pueden aparecer síntomas por trastornos compulsivos del comportamiento incluyendo casos de juego patológico, aumento de la libido, hipersexualidad, compra compulsiva o gasto, episodios de atracón y comer compulsivamente. Si estos síntomas aparecen se debe reducir la dosis o interrumpir el tratamiento gradualmente. Síndrome neuroléptico maligno: Se han reportado síntomas sugestivos de síndrome maligno neuroléptico con el retiro abrupto de la terapia dopaminérgica. Por lo tanto, se recomienda reducir el tratamiento. Pensamientos y conductas anormales: Se han reportado pensamientos y conductas anormales y éstos pueden consistir en una variedad de manifestaciones, incluyendo ideación paranoide, alucinaciones, confusión, conductas de tipo psicóticas, desorientación, agitación y conductas agresivas. Complicaciones fibróticas: Casos de fibrosis retroperitoneal, infiltrados pulmonares, derrame pleural, engrosamiento pleural, pericarditis y valvulopatía cardiaca, han sido reportados en algunos pacientes tratados con fármacos dopaminérgicos derivados de la ergotamina. Aunque estas complicaciones pueden desaparecer cuando se suspende la administración del fármaco, no siempre se produce la desaparición completa. Aunque estas reacciones adversas parecen estar relacionadas con la estructura ergotamínica de estos compuestos, se desconoce si hay otros agonistas dopaminérgicos no derivados de la ergotamina que también los produzcan. Aplicación de calor externo: No debe aplicarse calor externo (luz solar excesiva, compresas calientes y otras fuentes de calor, como el sauna o un baño caliente) en la zona del parche. Reacciones en el sitio de aplicación: Pueden producirse reacciones cutáneas en el lugar de aplicación, habitualmente leves o moderadas. Se recomienda rotar el lugar de aplicación cada día. Se debe evitar usar la misma zona antes de 14 días. Si aparecen reacciones en el lugar de aplicación que duren más de algunos días o que sean persistentes, si aumenta su intensidad o si la reacción cutánea se extiende fuera del lugar de aplicación, se debe realizar un estudio del balance riesgo-beneficio para el paciente. Si se produce un exantema cutáneo o irritación debido al sistema transdérmico, se debe evitar la exposición a la luz solar directa hasta que desaparezca completamente. La exposición podría provocar cambios de coloración cutánea. Se debe interrumpir el uso de NUBRENZA® si se observa una reacción cutánea generalizada asociada al uso de este medicamento. Sensibilidad a los sulfitos: NUBRENZA® contiene metabisulfito de sodio, un sulfito que puede causar reacciones alérgicas incluyendo síntomas anafilácticos, amenaza de vida o episodios asmáticos severos en pacientes susceptibles. Edema periférico: Como sucede con otros agonistas dopaminérgicos, la rotigotina se ha asociado con el desarrollo edema periférico en algunos pacientes con la enfermedad de Parkinson. Efectos sobre la capacidad para conducir y utilizar máquinas: La influencia de NUBRENZA® sobre la capacidad para conducir y utilizar máquinas es importante. Se debe informar a los pacientes en tratamiento con rotigotina que presenten somnolencia y/o episodios de inicio repentino de sueño que se abstengan de conducir o participar en actividades (p. ej., manejo de máquinas) en las que la reducción del estado de alerta pueda suponer un riesgo de lesión grave o muerte para ellos o para los demás, hasta que tales episodios recurrentes y la somnolencia hayan desaparecido.
Restricciones de uso durante el embarazo y la lactancia: Mujeres con potencial reproductivo/anticoncepción en hombres y mujeres: Las mujeres con potencial reproductivo deben utilizar la anticoncepción adecuada para evitar el embarazo durante el tratamiento con rotigotina. Embarazo: No hay datos suficientes del uso de NUBRENZA® en mujeres embarazadas. Los estudios realizados en animales no indican efectos teratógenos en ratas y conejos, pero se ha observado toxicidad embrionaria en ratas y ratones a dosis tóxicas para la madre. Se desconoce el riesgo potencial en seres humanos. Rotigotina no se debe usar durante el embarazo. Lactancia: Como rotigotina disminuye la secreción de prolactina en humanos, se espera inhibición de la lactancia. En los estudios con ratas se ha demostrado que rotigotina o sus metabolitos se excretan en la leche materna. Debido a la ausencia de datos en el ser humano, se debe interrumpir la lactancia. Fertilidad: Para información sobre los estudios de fertilidad, véase Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad.
Reacciones secundarias y adversas: Estudios clínicos: Panorama general: Las reacciones adversas (RA) reportadas en más del 10% de los pacientes con enfermedad de Parkinson tratados con NUBRENZA® son: Náuseas, mareo, somnolencia y reacciones en el sitio de aplicación. Al comienzo del tratamiento pueden presentarse reacciones adversas dopaminérgicas tales como náuseas y vómitos. Éstas son usualmente de leves a moderadas en intensidad y transitorias aun si el tratamiento se interrumpe. En los estudios en los que se rotó el lugar de aplicación como está indicado en el instructivo, la mayoría de las reacciones en el sitio de aplicación fueron leves o moderadas y limitadas a las zonas de aplicación. Listado de reacciones adversas: La tabla siguiente cubre las reacciones adversas al medicamento de todos los estudios en pacientes con enfermedad de Parkinson y la experiencia posterior a la comercialización.
Interacciones medicamentosas y de otro género: Antagonistas de la dopamina: Como rotigotina es un agonista dopaminérgico, se supone que los antagonistas dopaminérgicos como los neurolépticos (p. ej., las fenotiazinas, butirofenonas o tioxantenos) o metoclopramida pueden disminuir la eficacia de NUBRENZA®. Medicamentos sedantes: Debido a los posibles efectos aditivos, se deben tomar precauciones durante el tratamiento con sedantes u otros depresores del SNC (sistema nervioso central) (p. ej., benzodiazepinas, antipsicóticos o antidepresivos) o al tomar alcohol junto con rotigotina. L-dopa y carbidopa: La administración simultánea de L-dopa y carbidopa con rotigotina no afectó la farmacocinética de rotigotina, y la administración de rotigotina no afectó a la farmacocinética de L-dopa y carbidopa. La incidencia de algunos eventos adversos dopaminérgicos, como alucinaciones, discinesia y edema periférico generalmente es más alta cuando se administra en combinación con L-dopa. Domperidona: La administración simultánea de domperidona con rotigotina no tiene efecto en la farmacocinética de rotigotina. Anticonceptivos orales: La administración de rotigotina 3 mg/24 horas no afecta la farmacodinámica y farmacocinética de los anticonceptivos orales (0.03 mg etinilestradiol, 0.15 mg levonorgestrel). Omeprazol: La administración simultánea de 40 mg/día de omeprazol (inhibidor CYP2C19) no tiene efecto sobre la farmacocinética del estado de rotigotina (4 mg/24 horas).
Alteraciones en los resultados de pruebas de laboratorio: No aplicable.
Precauciones y relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: En los estudios de administración repetida y a largo plazo, los principales efectos estuvieron relacionados con los efectos farmacodinámicos derivados del agonismo de dopamina y el consecuente descenso de la secreción de prolactina. Después de la aplicación de una dosis única de rotigotina, se observó su unión a tejidos ricos en melanina (p. ej., los ojos) en la rata pigmentada y el mono, pero se eliminó lentamente durante el periodo de observación de 14 días. En un estudio con ratas albinas de 3 meses de duración, se observó al microscopio de transmisión la degeneración de la retina con una dosis equivalente a 2.8 veces la dosis máxima recomendada en humanos en mg/m2. El efecto fue más pronunciado en las ratas hembra. No se han realizado estudios adicionales para evaluar la causa patológica específica. No se observó degeneración retiniana durante la evaluación histopatológica de rutina de los ojos en ninguno de los estudios toxicológicos ni en las especies estudiadas. Se desconoce la relevancia de estos datos en el ser humano. En un estudio de carcinogénesis, las ratas macho desarrollaron tumores e hiperplasia de las células de Leydig. Aparecieron tumores malignos, predominantemente en el útero de las hembras tratadas con dosis medias y altas. Estas alteraciones son efectos conocidos tras el tratamiento durante toda la vida con agonistas dopaminérgicos en ratas y se evaluaron como no relevantes para el hombre. Los efectos de rotigotina sobre la reproducción se han estudiado en ratas, conejos y ratones. Rotigotina no fue teratogénico en ninguna de las tres especies, pero sí fue embriotóxica en ratas y ratones a dosis tóxicas para la madre. La rotigotina no influye en la fertilidad de las ratas macho, pero redujo claramente la fertilidad de las ratas y ratones hembras, debido a sus efectos sobre las concentraciones de prolactina, particularmente significativos en los roedores. Rotigotina no indujo mutaciones génicas en la prueba de Ames, pero se demostraron efectos en la prueba de linfoma de ratón in vitro con activación metabólica y efectos más débiles sin activación metabólica. Este efecto mutágeno podría atribuirse al efecto clastogénico de rotigotina, pero no se confirmó en la prueba de micronúcleos de ratón in vivo ni en la prueba de síntesis de ADN no programada (UDS) en la rata. Al ser más o menos paralelo al descenso del crecimiento relativo total de las células, este efecto puede estar relacionado con un efecto citotóxico del compuesto. Por tanto, se desconoce la trascendencia de una prueba de mutagénesis in vitro positiva. Estudios clínicos: Estudios clínicos en la enfermedad de Parkinson: La efectividad de NUBRENZA® en el tratamiento de los signos y síntomas de la enfermedad de Parkinson idiopática se evaluó en un programa de desarrollo farmacológico multinacional que consistió en cuatro estudios fundamentales, paralelos, aleatorizados, doble ciego y controlados con placebo y tres estudios que investigaron aspectos específicos de la enfermedad de Parkinson. Dos estudios fundamentales: (SP512, Parte I y SP513, Parte I) que investigaban la eficacia de NUBRENZA® en el tratamiento de los signos y síntomas de la enfermedad de Parkinson idiopática se realizaron en pacientes que no recibían tratamiento concomitante con un agonista dopaminérgico y que no habían recibido previamente tratamiento con L-dopa o que lo habían recibido durante ≤ 6 meses. El criterio de valoración principal se basó en la suma de la puntuación del componente de actividades de la vida diaria (ADL) (parte II), más el componente de la exploración motora (parte III) de la escala de puntuación unificada para la enfermedad de Parkinson (UPDRS). La eficacia se determinó según la respuesta del paciente al tratamiento en términos de respondedor y de mejoría absoluta, atendiendo a la suma de las puntuaciones de las secciones de ADL y exploración motora (parte II+III de la UPDRS). En el estudio doble ciego SP512, parte I, 177 pacientes recibieron rotigotina y 96 recibieron placebo. Se ajustó en cada paciente la dosis óptima de rotigotina o placebo, iniciando el tratamiento con una dosis de 2 mg/24 h, e incrementándola semanalmente en 2 mg/24 h, hasta un máximo de 6 mg/24 h la dosis óptima de cada paciente se mantuvo durante 6 meses. Al final del tratamiento de mantenimiento, en el 91% de los sujetos del grupo de rotigotina la dosis óptima fue la dosis máxima permitida, es decir, 6 mg/24 h. Se apreció una mejoría del 20% en el 48% de los sujetos tratados con rotigotina y en el 19% de los tratados con placebo (diferencia, 29% [IC95%] 18%; 39%, p < 0.0001). Con rotigotina, la media de la mejoría de la puntuación de la UPDRS (partes II+III) fue de -3.98 puntos (basal, 29.9 puntos), mientras que en el grupo tratado con placebo se observó un empeoramiento de 1.31 puntos (basal, 30.0 puntos). La diferencia fue de 5.28 puntos, estadísticamente significativa (p < 0.0001). En el estudio doble ciego SP513, parte I, 213 pacientes recibieron rotigotina, 227 recibieron ropinirol y 117 recibieron placebo. Se ajustó en cada paciente la dosis óptima de rotigotina, iniciando el tratamiento con una dosis de 2 mg/24 h, e incrementándola semanalmente en 2 mg/24 h, hasta un máximo de 8 mg/24 h a lo largo de 4 semanas. En el grupo de ropinirol se ajustó la dosis óptima hasta un máximo de 24 mg/día a lo largo de 13 semanas. En ambos grupos de pacientes, la dosis óptima se mantuvo durante 6 meses. Al final del tratamiento de mantenimiento, en el 92% de los sujetos del grupo de rotigotina la dosis óptima fue la dosis máxima permitida, es decir, 8 mg/24 h. Se apreció una mejoría del 20% en el 52% de los sujetos tratados con rotigotina, el 68% de los tratados con ropinirol y el 30% de los tratados con placebo (diferencia entre rotigotina y placebo: 21.7%; [IC95%]: 11.1%; 32.4%; diferencia entre ropinirol y placebo: 38.4%, [IC95%]: 28.1%; 48.6%; diferencia entre ropinirol y rotigotina: 16.6%; [IC95%]: 7.6%;.25.7%. La media de la mejoría en la puntuación de la UPDRS (partes II+III) fue de 6.83 puntos (basal: 33.2 puntos) en el grupo rotigotina, 10.78 puntos en el grupo ropinirol (basal: 32.2 puntos) y 2.33 puntos en el grupo de placebo (basal: 31.3 puntos). Todas las diferencias entre los tratamientos activos y el placebo fueron estadísticamente significativas. Se realizaron dos estudios fundamentales adicionales (SP650DB y SP515) en pacientes que recibían tratamiento concomitante con levodopa. El principal criterio de valoración fue la reducción del tiempo en "off" (horas). La eficacia se determinó según la respuesta del sujeto al tratamiento en términos de respondedor y de mejoría absoluta del tiempo pasado en "off". En el estudio doble ciego SP650B, 113 pacientes recibieron rotigotina hasta una dosis máxima de 8 mg/24 h, 109 pacientes recibieron rotigotina hasta una dosis máxima de 12 mg/24 h y 119 pacientes recibieron placebo. Los pacientes ajustaron su dosis óptima de rotigotina o placebo con incrementos semanales de 2 mg/24 h a partir de 4 mg/24 h. Los pacientes de cada grupo de tratamiento se mantuvieron con la dosis óptima durante 6 meses. Al terminar el tratamiento de mantenimiento se observó una mejoría al menos del 30% en el 57% y el 55% de los sujetos que recibieron 8 mg/24 h y 12 mg/24 h de rotigotina, respectivamente, y en el 34% de los sujetos tratados con placebo (diferencias del 22% y 21%, respectivamente, [IC95%] 10%; 35% y 8%; 33%, respectivamente, p < 0.001 para ambos grupos de rotigotina). Con rotigotina, la media de la reducción del tiempo en "off" fue de 2.7 y 2.1 horas, respectivamente, mientras que en el grupo tratado con placebo se observó una reducción de 0.9 horas. Las diferencias fueron estadísticamente significativas (p < 0.001 y p=0.003, respectivamente). En el estudio doble ciego SP515, 201 pacientes recibieron rotigotina, 200 recibieron pramipexol y 100 recibieron placebo. Los pacientes ajustaron su dosis óptima de rotigotina en incrementos semanales de 2 mg/24 h empezando con 4 mg/24 h hasta una dosis máxima de 16 mg/24 h. En el grupo pramipexol los pacientes recibieron 0.375 mg en la primera semana, 0.75 mg en la segunda semana y después se ajustaron la dosis en incrementos semanales de 0.75 mg hasta su dosis óptima hasta un máximo de 4.5 mg/día. Los pacientes de cada grupo de tratamiento se mantuvieron en el estudio durante 4 meses. Al terminar el tratamiento de mantenimiento se observó una mejoría al menos del 30% en el 60% de los casos tratados con rotigotina, el 67% de los sujetos tratados con pramipexol y el 35% de los tratados con placebo (diferencia de rotigotina frente a placebo: 25%; [IC95%] 13%; 36%, diferencia de pramipexol frente a placebo 32% IC95%: 21%; 43%, diferencia de pramipexol frente a rotigotina: 7%; [ICI95%]: -2%; 17%). La reducción media del tiempo en "off" fue de 2.5 horas en el grupo de rotigotina, 2.8 horas en el grupo pramipexol y 0.9 horas en el grupo placebo. Todas las diferencias entre los tratamientos activos y placebo fueron estadísticamente significativas. Se evaluaron los efectos de la rotigotina sobre aspectos específicos de la enfermedad de Parkinson en estudios adicionales (SP824, SP825 y SP889): En el estudio SP824, un estudio multicéntrico, multinacional, se ha estudiado la tolerabilidad para cambiar directamente de ropinirol, pramipexol o carbegolina a rotigotina parche transdérmico y sus efectos sobre los síntomas en pacientes con enfermedad de Parkinson idiopática. 116 pacientes fueron cambiados de la terapia oral previa para recibir hasta 8 mg/24 h de rotigotina, de entre ellos 47 habían sido tratados con ropinirol hasta 9 mg/ día, 47 habían sido tratados con pramipexol hasta 2 mg/ día y 22 habían sido tratados con carbegolina hasta 3 mg/ día. El cambio a rotigotina fue posible, con un ajuste de dosis menor (mediana: 2 mg/24 h) necesario sólo en 2 pacientes que cambiaban de ropinirol, en 5 pacientes de pramipexol y en 4 pacientes de carbegolina. Se observaron mejorías en las puntuaciones de la escala UPDRS parte I-IV. El perfil de seguridad no fue diferente del observado en estudios anteriores. En un estudio de asignación aleatoria, etiqueta abierta (SP825) en pacientes en etapas tempranas de la enfermedad de Parkinson, se asignaron de forma aleatoria a 25 pacientes al tratamiento con rotigotina y a 26 con ropinirol. En ambos grupos la dosis de tratamiento se ajustó hasta la dosis óptima o máxima de 8 mg/24 h o 9 mg /día, respectivamente. Ambos tratamientos mostraron mejorías en la función motora matutina y en el sueño. Los síntomas motores (UPDRS parte III) mejoraron en 6.3 ± 1.3 puntos en los pacientes tratados con rotigotina, y en 5.9 ± 1.3 puntos en el grupo de ropinirol tras 4 semanas de mantenimiento. EL sueño (PDSS) mejoró en 4.1 ± 13.8 puntos para los pacientes tratados con rotigotina, y en 2.5 ± 13.5 puntos para los pacientes tratados con ropinirol. El perfil de seguridad fue comparable a excepción de las reacciones en el sitio de aplicación. En los estudios SP824 y SP825, se demostró que la rotigotina y el ropinirol a dosis equivalente fueron comparables en eficacia. Se realizó otro estudio doble ciego multinacional (SP 889) en 287 pacientes con estadios tempranos o avanzados de enfermedad de Parkinson que tenían un control no satisfactorio de los síntomas motores en las primeras horas de la mañana. De estos pacientes, 81.5% estaban en tratamiento concomitante con levodopa, 190 pacientes recibían rotigotina y 97 placebo. Se ajustó la dosis de los pacientes a su dosis óptima de rotigotina o placebo en incrementos semanales de 2 mg/24 horas iniciando con 2 mg/24 horas hasta una dosis máxima de 16 mg/24 horas durante 8 semanas. Los pacientes de ambos grupos de tratamiento se mantuvieron con su dosis óptima durante 4 semanas. Las mediciones de resultados co-primarios fueron la función motora en las primeras horas de la mañana evaluada por medio de UPDRS parte III y trastornos del sueño nocturnos, evaluados por medio de la escala modificada del sueño en la enfermedad de Parkinson (PDSS-2). Al final del mantenimiento, la puntuación promedio en UPDRS parte III había mejorado 7.0 puntos en los pacientes tratados con rotigotina (basal 29.6), y 3.9 puntos en el grupo tratado con placebo (basal 32.0). Las mejorías en la puntuación total promedio de PDSS fueron 5.9 (rotigotina, basal 19.3) y 1.9 puntos (placebo, basal 20.5). Las diferencias del tratamiento en las variables co-primarias fueron estadísticamente significativas (p=0.0002 y p < 0.0001). En los análisis predefinidos de las mediciones secundarias y otras mediciones de resultados, se observaron mejorías marcadas desde el valor basal al final del mantenimiento con rotigotina en comparación con placebo: Puntuación de acinesia nocturna, distonía y calambres, escala de síntomas no motores de la enfermedad de Parkinson, inventario de depresión de Beck, escala de dolor Likert de 11 puntos, forma abreviada del cuestionario de la enfermedad de Parkinson, UPDRS parte II y UPDRS parte II + III. Se observó poco cambio en cualquiera de los grupos en el número de nicturias y UPDRS parte IV.
Dosis y vía de administración: NUBRENZA® se aplica una vez al día. Se debe aplicar el parche aproximadamente a la misma hora diariamente. El parche permanece sobre la piel durante 24 horas y deberá ser reemplazado por un parche nuevo en un sitio diferente de la aplicación anterior. Dosis: Las dosis recomendadas son dosis nominales. La dosificación en pacientes en las fases iniciales de la enfermedad de Parkinson: Debe iniciarse con una dosis diaria única 2 mg/24 horas e incrementos semanales posteriores de 2 mg/24 horas hasta alcanzar una dosis efectiva con una dosis máxima recomendada de 8 mg/24 horas. La dosis de 4 mg/24 horas puede ser efectiva en algunos pacientes. Para la mayoría de los pacientes la dosis efectiva se alcanza dentro de las primeras 3 a 4 semanas a dosis de 6 mg/24 horas u 8 mg/24 horas, respectivamente. La dosis máxima recomendada es de 8 mg/24 horas. Dosis en pacientes con enfermedad de Parkinson en fases avanzadas con fluctuaciones: Debe iniciarse con una dosis diaria única 4 mg/24 horas y tener incrementos semanales de 2 mg/24 horas hasta alcanzar una dosis efectiva hasta una dosis máxima de 16 mg/24 horas. Las dosis de 4 mg/24 horas o 6 mg/24 horas pueden ser efectivas en algunos pacientes. Para la mayoría de los pacientes la dosis efectiva se alcanza dentro de las primeras 3 a 7 semanas a dosis de 8 mg/24 horas, teniendo como dosis máxima 16 mg/24 horas. Para dosis más altas de 8 mg/24 horas pueden ser utilizados varios parches hasta la dosis final, por ejemplo 10 mg/24 horas pueden ser obtenidos combinando un parche de 6 mg/24 horas y uno de 4 mg/24 horas. Interrupción del tratamiento: NUBRENZA® debe suspenderse en forma gradual. La dosis diaria debe reducirse en fases de 2 mg/ 24 horas con una reducción de la dosis preferentemente cada tercer día hasta el retiro total de NUBRENZA®. Método de aplicación: El parche debe ser aplicado sobre la piel limpia, seca y sana en el abdomen, muslo, cadera, costado, hombro o brazo. Debe evitarse la aplicación en el mismo sitio dentro de los 14 días siguientes. No debe aplicarse el parche de NUBRENZA® si la piel esta roja, irritada o dañada. Uso y manejo: Cada parche está empacado en un sobre y debe aplicarse directamente después de que el sobre ha sido abierto. Una vez removida la mitad protectora se aplica la parte adherente del parche sobre la piel presionando firmemente. Posteriormente, se hace un doblez para liberar la otra mitad del parche de su protector. No debe tocarse la parte adherente del parche. Para que se adhiera firmemente a la piel, debe presionar firmemente sobre el parche con la palma de la mano durante 20 a 30 segundos. En caso de que el paciente olvide cambiar el parche a la hora usual del día, deberá aplicarse un parche nuevo por el tiempo restante para completar 24 horas. En caso de que el parche se despegue, debe aplicarse un parche nuevo por el tiempo restante para completar 24 horas de intervalo de dosis. El parche no debe cortarse en fragmentos. Después de su uso el parche aún contiene principio activo. Después de retirarlo, el parche se debe doblar a la mitad con el lado adhesivo hacia adentro de manera que la capa de la matriz no quede expuesta, colocar en el sobre original y después desechar fuera del alcance de los niños. Poblaciones especiales: Insuficiencia renal: No es necesario ajustar la dosis en pacientes con insuficiencia renal leve a severa, incluyendo a aquellos que requieren diálisis. Insuficiencia hepática: No se requiere el ajuste de la dosis en pacientes con insuficiencia hepática de leve a moderada. Rotigotina no ha sido investigada en pacientes con insuficiencia hepática severa. Niños y adolescentes: NUBRENZA® no se recomienda para su uso en niños y adolescentes debido a que aún no se cuenta con información de seguridad y eficacia en tales edades. Manifestaciones y manejo de la sobredosificación o ingesta accidental: Síntomas: Las reacciones adversas más probables serían las relacionadas con el perfil farmacodinámico de un agonista dopaminérgico, como náuseas, vómitos, hipotensión, movimientos involuntarios, alucinaciones, confusión, convulsiones y otros signos de estimulación dopaminérgica central. Tratamiento: No hay un antídoto conocido para la sobredosis de los agonistas dopaminérgicos. En caso de sospecha de sobredosis, se debe considerar la remoción del(los) parche(s). Después de retirar el(los) parche(s) el ingreso de fármaco se detiene y la concentración de rotigotina en el plasma disminuye rápidamente. El paciente debe ser supervisado estrechamente, incluyendo su frecuencia cardiaca, ritmo cardiaco y presión sanguínea. El tratamiento de la sobredosis puede requerir medidas de apoyo generales para mantener los signos vitales. No se esperaría que la diálisis tuviera beneficios, ya que la rotigotina no se elimina mediante la diálisis. Si es necesario interrumpir la rotigotina, esto debe hacerse gradualmente para evitar el síndrome maligno neuroléptico.
Presentaciones: Caja con 7, 14 o 28 parches de 2 mg/24 h, 4 mg/24 h, 6 mg/24 h u 8 mg/24 h.
Recomendaciones sobre almacenamiento: Consérvese a no más de 30°C.
Leyendas de protección: Léase el instructivo anexo. Mantener fuera de la vista y el alcance de los niños. Su venta requiere receta médica. No se use en el embarazoni lactancia. Literatura exclusiva para médicos. Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx
Nombre y domicilio del laboratorio: PRODUCTOS FARMACÉUTICOS, S. A. de C. V.
Km. 4.2 carretera a Pabellón de Hidalgo Rincón de Romos, 20420, Aguascalientes, México
Número de registro del medicamento: 249M2009, SSA IV