OFEV
BOEHRINGER PM
Denominación genérica: Nintedanib
Forma farmacéutica y formulación: Cada cápsula de 100 mg de OFEV® contiene: Nintedanib 100 mg. Cada cápsula de 150 mg de OFEV® contiene: Nintedanib 150 mg.Excipiente cbp 1 cápsula
Indicaciones terapéuticas: OFEV® está indicado para el tratamiento de la fibrosis pulmonar idiopática (FPI) en adultos. OFEV® está indicado para el tratamiento de la enfermedad pulmonar intersticial asociada a esclerosis sistémica (SSc-ILD).
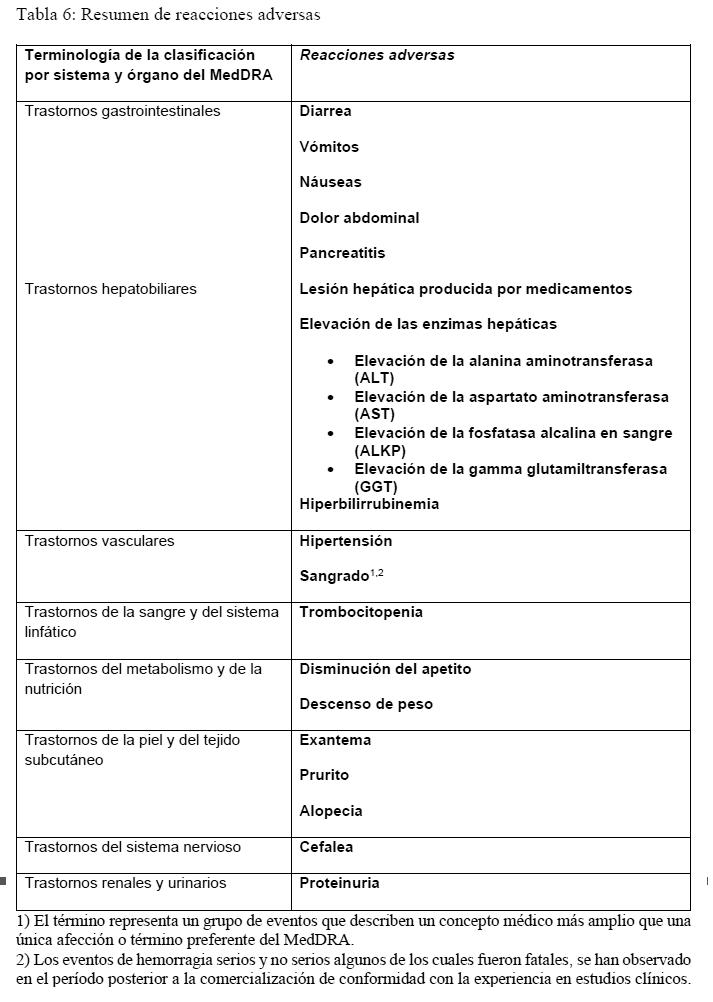
Farmacocinética y farmacodinamia: Mecanismo de acción: Nintedanib es un inhibidor de la cinasa de tirosina de molécula pequeña cuya acción comprende los receptores de factor del crecimiento derivado de plaquetas (PDGFR) a y b, los receptores del factor de crecimiento de fibroblastos (FGFR) 1-3 y los receptores del factor de crecimiento del endotelio vascular (VEGFR) 1-3. Además, nintedanib inhibe las cinasas de LcK, Lyn, Src y CSFIR. Nintedanib se une competitivamente al sitio de unión a ATP de de estas cinasas y bloquea las cascadas de señalización intracelular que, según se ha demostrado, están involucradas en la patogenia de la reestructuración del tejido fibrótico. Efectos farmacodinámicos: En estudios in vitro con células humanas, se ha demostrado que nintedanib inhibe procesos que se presume están involucrados en el inicio de la patogenia fibrótica, la liberación de los mediadores profibróticos respecto de las células monocíticas en sangre periférica y la polarización de macrófagos a macrófagos activados en forma alternativa. Se ha demostrado que nintedanib inhibe procesos fundamentales en la fibrosis orgánica, proliferación y migración de fibroblastos y transformación en el fenotipo de miofibroblastos activos, y secreción de la matriz extracelular. En estudios realizados con animales en modelos múltiples de FPI, SSc/SSc-ILD, RA-ILD (Enfermedad pulmonar intersticial asociada a artritis reumatoide) y otras fibrosis orgánicas, nintedanib ha presentado efectos antinflamatorios y antifibróticos en el pulmón, piel, corazón, riñón e hígado. Nintedanib también ejerció actividad vascular. Redujo la apoptosis de las células endoteliales microvasculares dérmicas y atenuó la reestructuración vascular pulmonar al reducir la proliferación de células de la musculatura lisa vascular, el espesor de las paredes de los vasos sanguíneos pulmonares y el porcentaje de vasos sanguíneos pulmonares ocluidos. Estudios Clínicos: Fibrosis pulmonar idiopática (FPI): La eficacia clínica de OFEV® ha sido estudiada en pacientes con FPI en dos estudios aleatorizados, doble ciego, controlados con placebo, de fase III, con idéntico diseño (INPULSIS-1 e INPULSIS-2). Los pacientes fueron aleatorizados en una proporción de 3:2 a recibir tratamiento con OFEV® 150 mg o placebo dos veces al día durante 52 semanas. El criterio de valoración primario fue la tasa anual de disminución de la capacidad vital forzada (CVF). Los criterios de valoración secundarios clave fueron el cambio respecto del nivel basal en el puntaje total del Cuestionario Respiratorio de Saint George (Saint George"s Respiratory Questionnaire, SGRQ) a las 52 semanas y el tiempo hasta la primera exacerbación aguda de la FPI. Tasa anual de reducción de la CVF: La tasa anual de disminución de la CVF (en ml) se redujo significativamente en los pacientes que recibieron OFEV® en comparación con los pacientes que recibieron placebo. El efecto del tratamiento fue concordante en ambos estudios. Véase la Tabla 1 para consultar los resultados de los estudios en forma individual y combinados.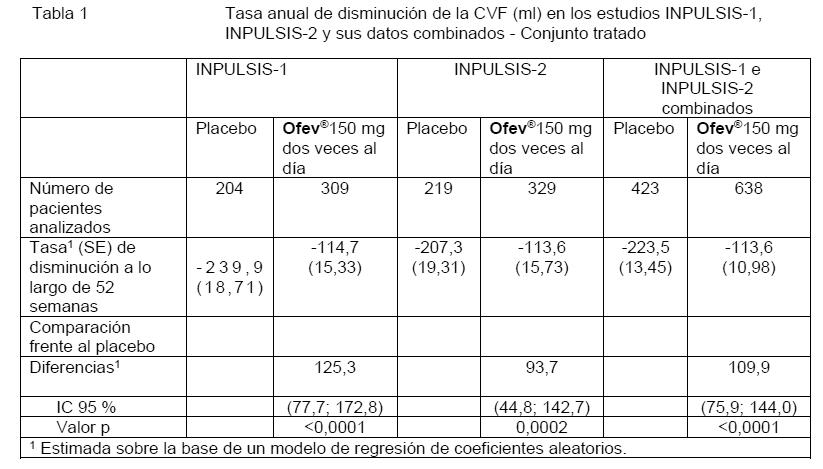
La robustez del efecto de OFEV® en la reducción de la tasa anual de disminución de la CVF fue confirmada en todos los análisis de sensibilidad previamente especificados. Asimismo, se observaron efectos similares en otros criterios de valoración de la función pulmonar, p. ej., cambio respecto del nivel basal en la CVF a la semana 52 y los análisis de respondedores por CVF confirman también los efectos de OFEV® en la ralentización de la progresión de la enfermedad. Véase la Figura 1 para la evolución del cambio respecto del nivel basal a lo largo del tiempo en ambos grupos de tratamiento, sobre la base del análisis de datos combinados de los estudios INPULSIS-1 e INPULSIS-2.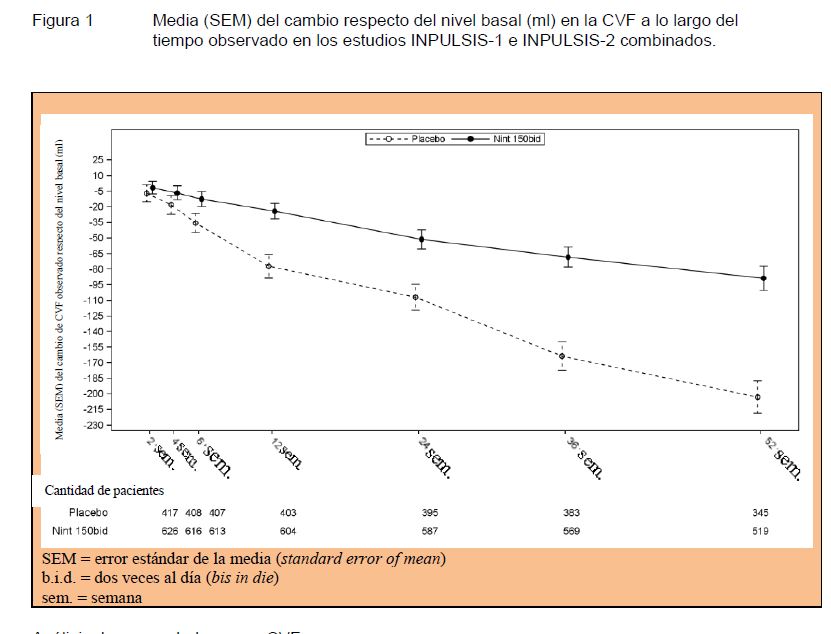
Análisis de respondedores por CVF: En ambos estudios INPULSIS, la proporción de respondedores por CVF, definidos como pacientes con un descenso absoluto en el % de CVF pronosticado de no más del 5% (un umbral indicativo del creciente riesgo de mortalidad en la FPI), fue significativamente más alto en el grupo de OFEV® en comparación con el grupo de placebo. Se observaron resultados similares en los análisis en los que se utilizó un umbral conservador del 10%. Véase la Tabla 2 para los resultados de los estudios en forma individual y combinados.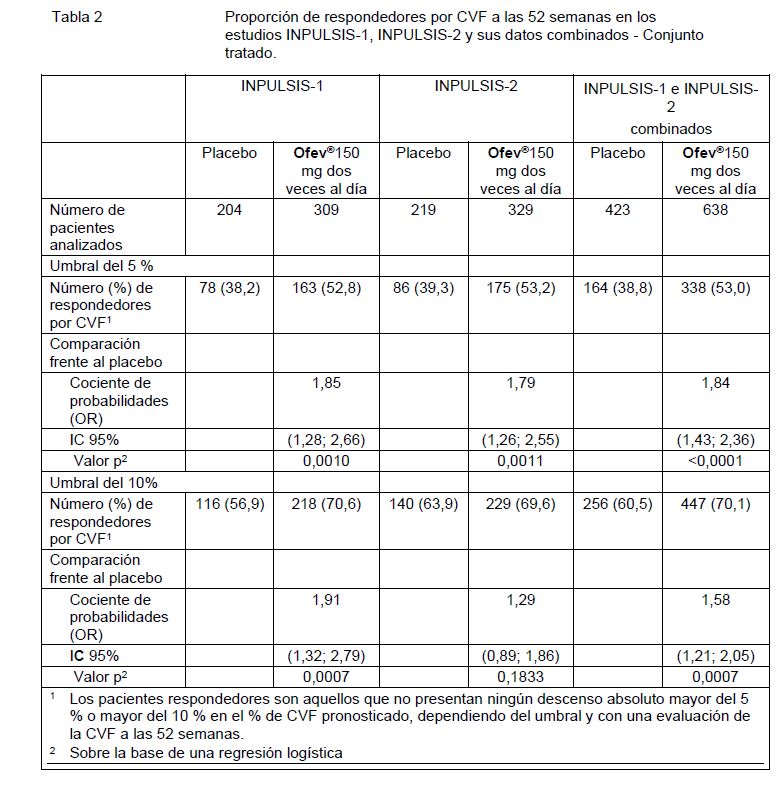
Tiempo hasta la progresión (≥ 10% descenso absoluto en el % de CVF pronosticado o muerte): En los dos estudios INPULSIS, el riesgo de progresión se redujo en una magnitud estadísticamente significativa en los pacientes tratados con OFEV® en comparación con aquellos tratados con placebo. En el análisis combinado, el valor de HR fue 0,60, lo que indicó una reducción del 40% en el riesgo de progresión para los pacientes tratados con OFEV® en comparación con aquellos tratados con placebo; ver Tabla 3.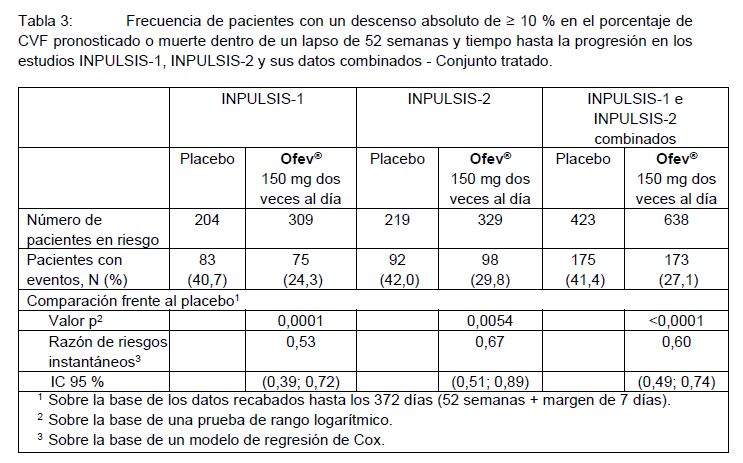
Cambio respecto del nivel basal en el puntaje total del SGRQ a la semana 52: El puntaje total del Cuestionario Respiratorio de St. George (St. George"s Respiratory Questionnaire, SGRQ), que mide la calidad de vida relacionada con la salud (health-related quality of life, HRQoL), se analizó a las 52 semanas. En el estudio INPULSIS-2, los pacientes que recibieron placebo tuvieron un mayor incremento respecto del nivel basal en el puntaje total del SGRQ en comparación con los pacientes que recibieron OFEV® 150 mg dos veces al día. El deterioro de la HRQoL fue menor en el grupo tratado con OFEV®; la diferencia entre los grupos de tratamiento fue estadísticamente significativa (-2,69; IC 95%: -4,95, -0,43; p = 0,0197). En el estudio INPULSIS-1, el incremento respecto del nivel basal en el puntaje total del SGRQ en la semana 52 fue comparable entre OFEV® y placebo (diferencia entre los grupos de tratamiento: -0,05, IC 95%: -2,50, 2,40; p = 0,9657). En el análisis combinado de los estudios INPULSIS, la media estimada del cambio entre el nivel basal y la semana 52 en el puntaje total de SGRQ fue menor en el grupo tratado con OFEV® (3,53) que en el grupo tratado con placebo (4,96), con una diferencia entre los grupos de tratamiento de - 1,43 (IC 95%: -3,09, 0,23; p = 0,0923). En conjunto, el efecto de OFEV® sobre la calidad de vida relacionada con la salud medida por el puntaje total de SGRQ es modesto, lo que indica un menor grado de empeoramiento en comparación con el placebo. Tiempo hasta la primera exacerbación aguda de la FPI: En el estudio INPULSIS-2, el riesgo de la primera exacerbación aguda de la FPI a lo largo de 52 semanas se redujo significativamente en los pacientes que recibieron OFEV® en comparación con aquellos que recibieron placebo; en el estudio INPULSIS-1, no hubo ninguna diferencia entre los grupos de tratamiento. En el análisis combinado de los estudios INPULSIS, se observó un riesgo numéricamente menor de una primera exacerbación aguda en los pacientes que recibieron OFEV® en comparación con aquellos que recibieron placebo. Véase la Tabla 4 para analizar los resultados de los estudios en forma individual y combinados.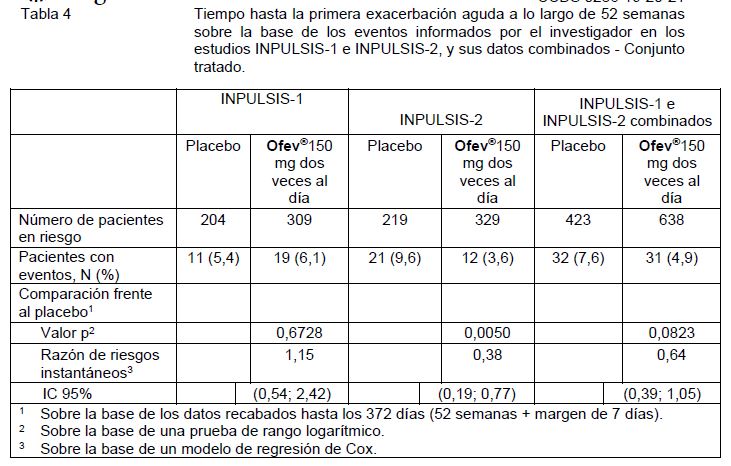
Todos los eventos adversos de exacerbación aguda de la FPI informados por el investigador fueron adjudicados por un comité de adjudicación con cegamiento. Se llevó a cabo un análisis de sensibilidad previamente especificado del tiempo hasta la primera exacerbación aguda de la FPI "confirmada" o "sospechada" sobre la base de los datos combinados. La frecuencia de pacientes con al menos 1 exacerbación adjudicada producida dentro de las 52 semanas fue más baja en el grupo de OFEV® (1,9% de los pacientes) que en el grupo de placebo (5,7% de los pacientes). El análisis del tiempo hasta el evento de los eventos de exacerbación adjudicados realizado sobre la base de los datos combinados arrojó una HR de 0,32 (IC 95% 0,16, 0,65; p = 0,0010). Esto indica que el riesgo de tener una primera exacerbación aguda de la FPI fue más bajo de manera estadísticamente significativa en el grupo tratado con OFEV® que en el grupo tratado con placebo en cualquiera de los momentos de medición. Análisis de la supervivencia: En el análisis combinado previamente especificado de los datos de supervivencia de los estudios INPULSIS, la mortalidad general a lo largo de 52 semanas fue más baja en el grupo tratado con OFEV® (5,5%) en comparación con el grupo tratado con placebo (7,8%). El análisis del tiempo hasta la muerte arrojó una HR de 0,70 (IC 95% 0,43, 1,12; p = 0,1399). Los resultados de todos los criterios de valoración de supervivencia (como la mortalidad durante el tratamiento y la mortalidad por causas respiratorias) evidenciaron una diferencia numérica sistemática a favor de OFEV® (ver Tabla 5).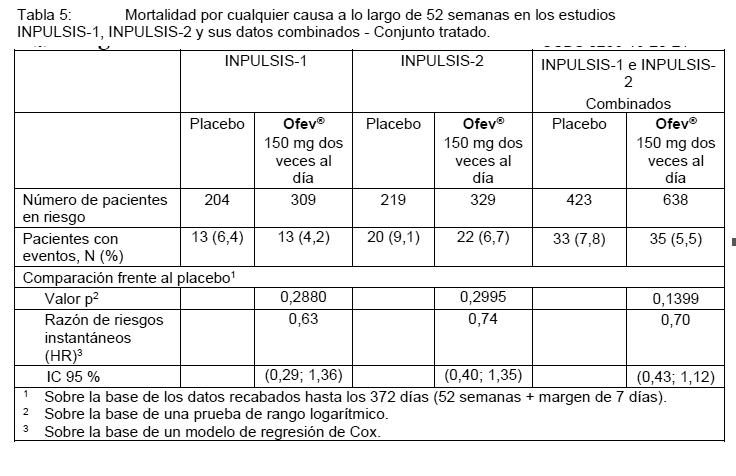
Evidencia de aval de los resultados del estudio de fase II (1199.30) con OFEV® 150 mg dos veces al día: El estudio aleatorizado, doble ciego, controlado con placebo, de búsqueda de dosis, de fase II en el que se incluyó un grupo de dosis de OFEV® de 150 mg dos veces por día aportó evidencia adicional de la eficacia.El criterio de valoración primario, la tasa de disminución de la CVF a lo largo de 52 semanas, registró su valor más bajo en el grupo de tratamiento con OFEV® (-0,060 L/año, N=84) que en el grupo de tratamiento con placebo (-0,190 L/año, N=83). La diferencia estimada entre los grupos de tratamiento fue 0,131 L/año (IC 95% 0,027, 0,235). Esta diferencia entre los grupos de tratamiento alcanzó la significancia estadística nominal (p = 0,0136). La media estimada del cambio respecto del nivel basal en el puntaje total del SGRQ a las 52 semanas fue 5,46 para el placebo, lo que indica un empeoramiento de la calidad de vida relacionada con la salud y de -0,66 para nintedanib, lo cual indica una calidad de vida relacionada con la salud estable. La diferencia media estimada para OFEV® en comparación con el placebo fue -6,12 (IC 95%: -10,57, -1,67; p = 0,0071). El número de pacientes con exacerbaciones agudas de la FPI a lo largo de 52 semanas fue más bajo en el grupo de OFEV® (2,3%, N = 86) en comparación con el grupo de placebo (13,8%, N = 87). La tasa de riesgos instantáneos (HR) estimada de OFEV® frente al placebo fue 0,16 (IC 95% 0,04, 0,71; p = 0,0054). Tratamiento a largo plazo con OFEV® en pacientes con FPI (INPULSIS-ON): Un estudio de extensión abierto de OFEV® incluyó 734 pacientes con FPI. Algunos pacientes fueron tratados con OFEV® durante más de 5 años. Los pacientes que completaron el periodo de tratamiento de 52 semanas en un estudio INPULSIS recibieron tratamiento abierto con OFEV® en el estudio de extensión INPULSIS-ON. La mediana del tiempo de exposición para los pacientes tratados con OFEV® en ambos estudios, INPULSIS e INPULSIS®-ON, fue de 44,7 meses (rango 11,9-68,3). La tasa anual ajustada de disminución de la CVF a lo largo de 192 semanas fue -135,1 (5,8) ml/año en todos los pacientes tratados y fue concordante con la tasa anual de disminución de la CVF en los pacientes tratados con OFEV® en los estudios INPULSIS de fase III (-113,6 ml por año). El perfil de eventos adversos de OFEV® en INPULSIS®-ON fue similar al de los estudios de fase III INPULSIS. Pacientes con FPI con deterioro de la función pulmonar avanzado (INSTAGE): En un estudio doble ciego, aleatorizado, de grupos paralelos se evaluó la eficacia y la seguridad de OFEV® coadministrado con sildenafil oral, en comparación con el tratamiento con OFEV® solo, en 273 pacientes con FPI y deterioro de la función pulmonar avanzado (DLCO < 35 % estimado) durante 24 semanas. La disminución de la CVF en los pacientes tratados OFEV® solo fue concordante con la disminución de la CVF en los pacientes con menor avance de la enfermedad y tratados con OFEV® en los estudios de fase III INPULSIS. La adición de sildenafil a OFEV® no brindó un beneficio significativo en términos de calidad de vida vs. la monoterapia de OFEV®. El perfil de seguridad y tolerabilidad de OFEV® en los pacientes con IPF con deterioro de la función pulmonar avanzado fue concordante con el observado en los estudios de fase III INPULSIS. El perfil de eventos adversos de la combinación de OFEV® y sildenafil fue coherente con el perfil de seguridad establecido de cada componente, sin aumento de los eventos adversos serios o mortales en comparación con la monoterapia de OFEV®. Datos adicionales del estudio clínico de fase IV INJOURNEY con OFEV® 150 mg dos veces al día y agregado de pirfenidona: Se investigó el tratamiento concomitante con OFEV® y pirfenidona en un estudio clínico abierto, aleatorizado, exploratorio de 150 mg de OFEV® dos veces al día más pirfenidona (con ajuste de dosis a 801 mg tres veces al día) en comparación con monoterapia de OFEV® 150 mg dos veces al día, en un total de 105 pacientes aleatorizados durante 12 semanas. El criterio de valoración primario fue el porcentaje de pacientes con eventos adversos gastrointestinales desde el inicio del estudio hasta la semana 12. Los eventos adversos gastrointestinales fueron frecuentes y coherentes con el perfil de seguridad establecido de cada componente. Los eventos adversos más frecuentes fueron diarrea, náuseas y vómitos, los cuales fueron informados por 20 (37,7%) versus 16 (31,4%), 22 (41,5%) versus 6 (11,8%) y 15 (28,3%) versus 6 (11,8%) pacientes tratados con la combinación nintedanib más pirfenidona en comparación con la monoterapia de OFEV®, respectivamente. La media (SE) de cambios absolutos desde el inicio en la CVF a la semana 12 fue de -13,3 (17,4) ml en los pacientes tratados con nintedanib más pirfenidona (n=48), en comparación con -40,9 (31,4) ml en los pacientes tratados con monoterapia de nintedanib (n=44). Enfermedad pulmonar intersticial asociada a esclerosis sistémica (SSc-ILD): Se ha estudiado la eficacia clínica de OFEV® en pacientes con SSc-ILD en un estudio doble ciego, aleatorizado, comparativo con placebo, de fase III (SENSCIS). Los pacientes fueron diagnosticados con SSc-ILD sobre la base de los o de clasificación para la SSc establecidos en 2013 por el Colegio de Reumatología de Estados Unidos (American College of Rheumatology)/Liga Europea contra el Reumatismo (European League Against Rheumatism) y de una tomografía computada de tórax de alta resolución (HRCT) realizada dentro de los 12 meses previos. Un total de 580 pacientes fueron aleatorizados (1:1) a recibir OFEV® 150 mg bid o placebo equivalente durante al menos 52 semanas, de los cuales 576 pacientes recibieron tratamiento. La aleatorización se estratificó de acuerdo con el estado de Anticuerpos anti-Topoisomerasa (ATA). Cada paciente permaneció en tratamiento en un estudio ciego durante 100 semanas (mediana de exposición de OFEV® 15,4 meses; media de exposición de OFEV® 14,5 meses). El objetivo primario fue la tasa anual de disminución de la capacidad vital forzada (CVF) a lo largo de 52 semanas. Los objetivos secundarios clave fueron el cambio absoluto respecto del nivel inicial en el puntaje de piel de Rodnan modificado (mRSS) en la semana 52 y el cambio absoluto respecto del nivel inicial en el puntaje total del Cuestionario Respiratorio St. George (SGRQ) en la semana 52. En la población general, el 75,2% de los pacientes eran de sexo femenino. La media de edad (desvío estándar [SD, Mín.-Máx.]) era de 54,0 (12,2; 20-79) años. En general, el 51,9% de los pacientes presentaba esclerosis sistémica cutánea (SSc) difusa y el 48,1% padecía SSc cutánea limitada. El tiempo promedio (SD) desde la primera aparición de un síntoma no asociado a la enfermedad de Raynaud fue 3,49 (1,7) años. El 49,0% de los pacientes permanecieron en tratamiento estable con micofenolato en el nivel inicial. El perfil de seguridad en los pacientes tratados con o sin micofenolato en el nivel inicial fue similar. Tasa anual de disminución de la CVF: La tasa anual de disminución de la CVF (en ml) a lo largo de 52 semanas se redujo significativamente en 41,0 ml en los pacientes que recibieron OFEV®, en comparación con los pacientes que recibieron placebo (Tabla 11), lo que corresponde a un efecto relativo del tratamiento del 43,8%.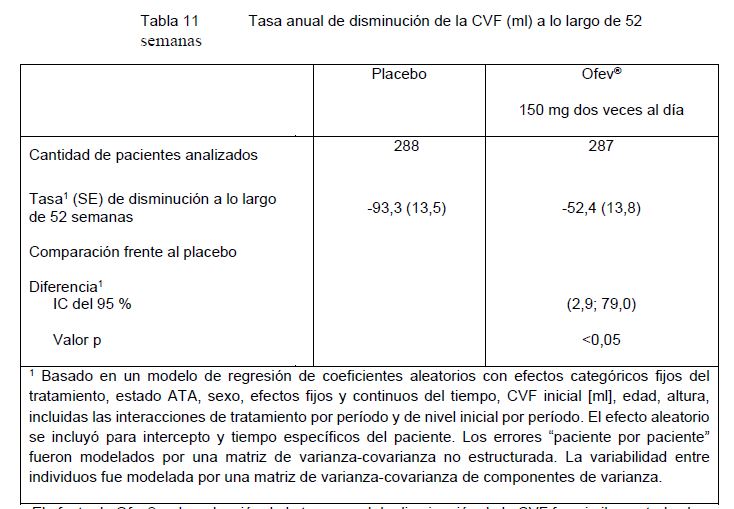
El efecto de OFEV® en la reducción de la tasa anual de disminución de la CVF fue similar en todos los análisis de sensibilidad previamente especificados, y no se detectó heterogeneidad en los subgrupos prespecificados (por ej, por edad, sexo y uso de micofenolato). Asimismo, se observaron efectos similares en otros objetivos de la función pulmonar, p. ej., cambio absoluto respecto del nivel inicial en la CVF en ml a la semana 52 (Figura 3 y Tabla 12) y tasa de disminución de la CVF en porcentaje previsto a lo largo de 52 semanas (Tabla 13) que confirman también los efectos de OFEV® en la ralentización de la progresión de SSc-ILD. Además, menos pacientes del grupo tratado con OFEV® tuvieron una disminución absoluta de la CVF > 5 % previsto (20,6 % en el grupo tratado con OFEV® frente al 28,5 % en el grupo tratado con placebo, OR = 0,65; P = 0,0287). La disminución relativa de la CVF en ml > 10% fue similar entre ambos grupos (16,7% en el grupo de OFEV® frente al 18,1% en el grupo placebo, OR = 0,91, p = 0,6842). En estos análisis, los valores faltantes de CVF en la semana 52 se imputaron con el peor valor del paciente durante el tratamiento. Un análisis exploratorio de datos de hasta 100 semanas (duración máxima del tratamiento en SENSCIS) sugirió que el efecto de OFEV® durante el tratamiento en la ralentización de la progresión de SSc-ILD perduró después de 52 semanas.
FIGURA 3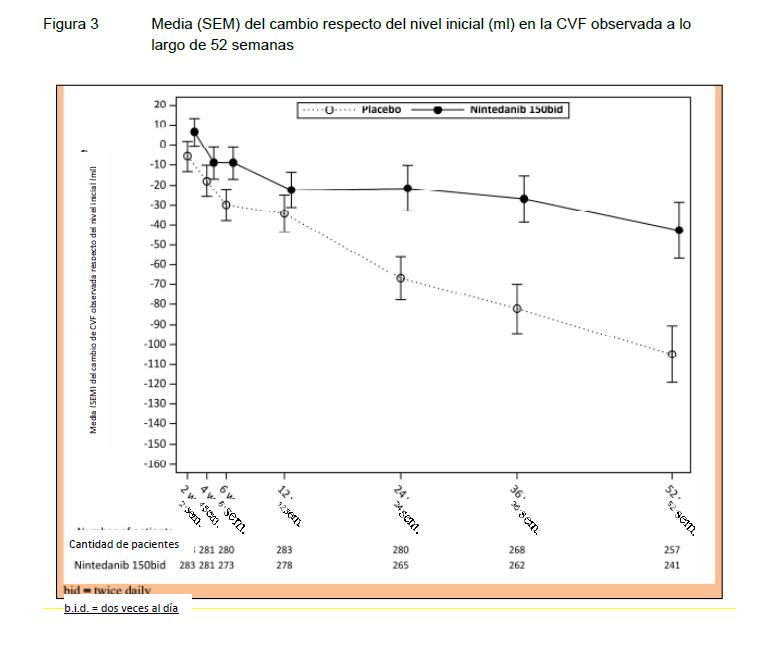
Cambio respecto del nivel inicial en el puntaje de piel de Rodnan modificado (mRSS) en la semana 52: El cambio absoluto respecto del nivel inicial de la media ajustada en mRSS en la semana 52 fue similar entre el grupo tratado con OFEV® (-2,17 (IC del 95% -2,69, -1,65)) y el grupo tratado con placebo (-1,96 (IC 95% -2,48, -1,45)). La diferencia media ajustada entre los grupos de tratamiento fue -0,21 (IC 95% -0,94, 0,53; p = 0,5785). Cambio respecto del nivel inicial en el puntaje total del Cuestionario Respiratorio St. George (SGRQ) en la semana 52: El cambio absoluto respecto del nivel inicial de la media ajustada en el puntaje total del SGRQ en la semana 52 fue similar entre el grupo tratado con OFEV® (0,81 (IC 95% -0,92, 2,55)) y el grupo tratado con placebo (-0,88 (IC 95% -2,58, 0,82)). La diferencia media ajustada entre los grupos de tratamiento fue 1,69 (IC 95% CI -0,73, 4,12; p = 0,1711). Análisis de la supervivencia: La mortalidad a lo largo de todo el estudio fue similar entre el grupo tratado con OFEV® (N = 10; 3,5 %) y el grupo tratado con placebo (N = 9; 3,1%). El análisis del tiempo hasta la muerte durante todo el estudio arrojó una HR de 1,16 (IC 95% 0,47, 2,84; p = 0,7535). Efecto sobre el intervalo QT: Se efectuaron y se analizaron mediciones de QT/QTc a partir de un estudio específico en el cual se comparó la monoterapia de nintedanib frente a la monoterapia de sunitinib en pacientes con carcinoma renal. En este estudio, dosis únicas orales de 200 mg de nintedanib y dosis múltiples orales de 200 mg de nintedanib administradas dos veces al día durante 15 días no prolongaron el intervalo QTcF. Población pediátrica: No se han llevado a cabo estudios clínicos en niños ni adolescentes. Farmacocinética: La farmacocinética de nintedanib puede considerarse lineal en relación con el tiempo (es decir, los datos de las dosis únicas pueden extrapolarse a datos de dosis múltiples). La acumulación observada tras la administración de dosis múltiples fue de 1,04 veces para la Cmáx y de 1,38 veces para el AUCt. Las concentraciones valle de nintedanib se mantuvieron estables durante más de un año. Absorción: Nintedanib alcanzó las concentraciones plasmáticas máximas aproximadamente 2-4 horas después de la administración por vía oral como cápsulas de gelatina blanda en estado posprandial (rango: 0,5-8 horas. La biodisponibilidad absoluta de una dosis de 100 mg fue 4,69% (IC 90%: 3,615-6,078) en voluntarios sanos. La absorción y la biodisponibilidad se ven reducidas por los efectos de los transportadores y por un grado sustancial de metabolismo de primer paso. La proporcionalidad a la dosis se demostró a través del incremento de la exposición a nintedanib (rango de dosis de 50-450 mg una vez al día y de 150-300 mg dos veces al día). Las concentraciones plasmáticas en estado de equilibrio dinámico se lograron dentro de un lapso de administración de una semana como máximo. Tras la ingesta de alimentos, la exposición a nintedanib se incrementó aproximadamente un 20% en comparación con la administración en ayunas (IC: 95,3-152,5%) y la absorción fue más lenta (mediana de tmáx; en ayunas: 2,00 horas; en estado posprandial: 3,98 h). Distribución: Nintedanib sigue una cinética de disposición como mínimo bifásica. Tras la infusión intravenosa, se observó un importante volumen de distribución (Vss: 1050 L, 45,0% gCV). El grado de unión a las proteínas de nintedanib observado in vitro en el plasma humano fue elevado, con una fracción ligada del 97,8%. Se considera que la albúmina sérica es la principal proteína de unión. Nintedanib se distribuye preferentemente en el plasma, con una relación sangre: plasma de 0,869. Metabolismo/Biotransformación: La reacción metabólica prevalente en el caso de nintedanib es la escisión hidrolítica por esterasas, que conduce a la formación de la fracción ácido libre BIBF 1202. BIBF 1202 luego es glucuronizado por las enzimas UGT (a saber, UGT 1A1, UGT 1A7, UGT 1A8 y UGT 1A10) con la consecuente transformación en el BIBF 1202 glucurónido. Sólo un grado mínimo de la biotransformación de nintedanib estuvo relacionada con las vías del CYP, siendo CYP 3A4 la enzima predominante en dicho proceso. El principal metabolito dependiente de CYP no pudo ser detectado en el plasma en el estudio de absorción, distribución, metabolismo y eliminación (ADME) en humanos. In vitro, el metabolismo dependiente de CYP representó aproximadamente un 5% en comparación con alrededor de un 25% en el caso de la escisión de ésteres. Eliminación: La depuración plasmática total tras la administración por infusión intravenosa fue elevada (CL: 1390 ml/min, 28,8% gCV). La eliminación urinaria del principio activo inalterado dentro de las 48 horas fue de aproximadamente el 0,05% de la dosis (gCV 31,5%) tras la administración por vía oral, y de aproximadamente el 1,4% de la dosis (gCV 24,2%) tras la administración por vía intravenosa; la depuración renal fue 20 ml/min (gCV 32,6%). La principal vía de eliminación de la radioactividad relacionada con el fármaco tras la administración por vía oral de [14C] nintedanib fue la excreción fecal/biliar (93,4% de la dosis, gCV 2,61%). La contribución de la eliminación renal a la depuración total fue baja (0,649% de la dosis, gCV 26,3%). La recuperación total se consideró completa (superior al 90%) dentro de los 4 días subsiguientes a la administración. La vida media terminal de nintedanib fue de entre 10 y 15 h (gCV % aprox. 50%. Relación exposición-respuesta: Los análisis de exposición-respuesta de los pacientes con FPI y SSc-ILD indicaron una relación pseudo Emáx entre la exposición en el rango observado en la Fase II y III y la tasa de descenso anual de la CVF con un EC50 de alrededor de 3-5 ng/ml (errores estándar relativos: 55%). Para fines comparativos, la mediana de las concentraciones valle de nintedanib observadas para 150 mg bid de OFEV® fue de aproximadamente 10 ng/ml. En lo que respecta a la seguridad, aparentemente hubo una relación débil entre la exposición plasmática a nintedanib y las elevaciones de los niveles de ALT y/o AST. La dosis real administrada podría ser un mejor predictor del riesgo de desarrollar diarrea de cualquier intensidad, incluso aunque no se pueda descartar la exposición plasmática como un factor de riesgo determinante (véase la sección Advertencias y Precauciones generales). Factores intrínsecos y extrínsecos-poblaciones especiales: Las propiedades farmacocinéticas de nintedanib fueron similares en los voluntarios sanos, en los pacientes con FPI pacientes con SSc-ILD y en los pacientes oncológicos. Sobre la base de los resultados de los análisis de farmacocinética poblacional y las investigaciones descriptivas, la exposición a nintedanib no se vio influenciada por el sexo (con corrección para peso corporal), la existencia de un deterioro renal leve o moderado (estimado sobre la base de la depuración de creatinina), la presencia de metástasis hepáticas, el puntaje de estado funcional ECOG, el consumo de alcohol ni el genotipo de P-gp. Los análisis de farmacocinética poblacional indicaron efectos moderados sobre la exposición a nintedanib dependientes de la edad, el peso corporal y la raza (ver a continuación). Sobre la base de la elevada variabilidad entre individuos de la exposición que se observó en los estudios clínicos, estos efectos no se consideran clínicamente relevantes (véase la sección Advertencias y precauciones generales). Edad: La exposición a nintedanib se incrementó en forma lineal en función de la edad. Los valores de AUCt,ss evidenciaron una reducción del 16% para un paciente de 45 años de edad (percentilo 5) y se incrementaron a razón de un 13% para un paciente de 76 años de edad (percentilo 95) respecto de un paciente con una mediana de edad de 62 años. El rango de edad cubierto por el análisis fue de 29 a 85 años; aproximadamente el 5% de la población fue mayor de 75 años. No se han efectuado estudios en poblaciones pediátricas. Peso corporal: Se observó una correlación inversa entre el peso corporal y la exposición a nintedanib. Los valores de AUCt,ss se incrementaron a razón de un 25% para un paciente de 50 kg (percentilo 5) y se redujeron a razón de un 19% para un paciente de 100 kg (percentilo 95) respecto de un paciente con una mediana de peso de 71,5 kg. Raza: La población expuesta a nintedanib fue 33-50% más alta en los pacientes procedentes de China, Taiwán e India y un 16% más alta en los pacientes japoneses, en tanto que fue 16-22% más baja en los pacientes de Corea, en comparación con los caucásicos (con corrección para peso corporal). Los datos obtenidos a partir de sujetos de raza negra fueron muy limitados, pero se ubicaron dentro del mismo rango que aquellos de los sujetos caucásicos. Insuficiencia hepática: En un estudio específico de dosis única de fase I en el que se tomó como blanco a sujetos sanos, la exposición al nintedanib, considerando tanto la Cmáx como el AUC, fue 2,2 veces mayor en voluntarios con insuficiencia hepática leve (Child Pugh A; IC 90% de la Cmáx: 1,3-3,7; IC 90% del AUC: 1,2-3,8 respectivamente). En voluntarios con insuficiencia hepática moderada (Child Pugh B), fue 7,6 veces mayor en términos de la Cmáx (IC 90%: 4,4-13,2) y 8,7 veces mayor en términos del AUC (IC 90%: 5,7-13,1), respectivamente, en comparación con pacientes sanos. No se estudiaron sujetos con insuficiencia hepática grave (Child Pugh C). Tratamiento concomitante con pirfenidona: Se estudió el tratamiento concomitante de OFEV® con pirfenidona en un estudio exclusivo de farmacocinética realizado en pacientes con FPI. El grupo 1 recibió una única dosis de 150 mg de OFEV® antes y después del ajuste ascendente de la dosis a 801 mg de pirfenidona tres veces al día en estado de equilibrio dinámico. El grupo 2 recibió tratamiento con 801 mg de pirfenidona tres veces al día en estado de equilibrio dinámico y se realizó una determinación del perfil farmacocinético antes y después de, al menos, 7 días de tratamiento concomitante con 150 mg de OFEV® dos veces al día. En el grupo 1, las proporciones ajustadas de la media geométrica (intervalo de confianza (IC) del 90%) fueron 93% (57% - 151%) y 96% (70% - 131%) para la Cmáx y AUC0-tz de nintedanib, respectivamente (n=12). En el grupo 2, las proporciones ajustadas de la media geométrica (IC del 90%) fueron 97% (86% - 110%) y 95% (86% - 106%) para la Cmáx,ss y AUCt,ss de pirfenidona, respectivamente (n=12). En base a estos resultados, no hay evidencia de la existencia de interacciones medicamentosas farmacocinéticas relevantes entre nintedanib y pirfenidona cuando estos fármacos se administran como tratamiento combinado. Tratamiento concomitante con bosentán: Se investigó el tratamiento concomitante de OFEV® con bosentán en un estudio exclusivo de farmacocinética realizado en voluntarios sanos. Los sujetos recibieron una única dosis de 150 mg de OFEV® antes y después de dosis múltiples de 125 mg de bosentán dos veces al día en estado de equilibrio dinámico. Las proporciones ajustadas de la media geométrica (intervalo de confianza (IC) del 90%) fueron 103% (86%-124%) y 99% (91%-107%) para la Cmáx y AUC0-tz de nintedanib, respectivamente (n=13), lo cual indica que la coadministración de nintedanib y bosentán no alteró la farmacocinética de nintedanib. Potencial de interacciones medicamentosas: Metabolismo: No es dable esperar que se produzcan interacciones medicamentosas entre nintedanib y los sustratos del CYP, los inhibidores del CYP o los inductores del CYP, ya que nintedanib, BIBF 1202 y el glucurónido BIBF 1202 no evidenciaron efectos de inhibición ni de inducción de las enzimas del CYP en los ensayos preclínicos y nintedanib no fue metabolizado en un grado relevante por las enzimas del CYP. Transporte: Nintedanib es un sustrato de la P-gp. Para el potencial de interacción de nintedanib con este transportador, véase la sección Interacciones. Se ha comprobado que nintedanib no es un sustrato ni un inhibidor de OATP-1B1, OATP-1B3, OATP-2B1, OCT-2 o MRP-2 in vitro. Nintedanib tampoco fue un sustrato de la proteína de resistencia al cáncer de mama (breast cancer resistance protein, BCRP). Sólo se observó un débil potencial inhibidor sobre OCT-1, BCRP y P-gp in vitro, el cual se considera de escasa relevancia clínica. Lo mismo aplica a nintedanib en cuanto a ser un sustrato de OCT-1.
Contraindicaciones: OFEV® está contraindicado en pacientes con hipersensibilidad conocida a nintedanib, el cacahuate o la soya, o a cualquiera de los excipientes (Véase la sección Forma farmacéutica y Formulación). OFEV® está contraindicado durante el embarazo (véanse las secciones Embarazo, lactancia y Fertilidad, Toxicología).
Precauciones generales: Trastornos gastrointestinales: • Diarrea: En los estudios clínicos (véase la sección Estudios clínicos), la diarrea fue el evento gastrointestinal informado con mayor frecuencia. En la mayoría de los pacientes, el evento fue de intensidad leve a moderada y se produjo dentro de los primeros 3 meses de tratamiento. En los estudios INPULSIS realizados en pacientes con FPI, se informó diarrea en el 62,4% versus el 18,4% de los pacientes tratados con OFEV® y placebo, respectivamente. La diarrea condujo a una reducción de la dosis de OFEV®en el 10,7% de los pacientes y a la interrupción de OFEV® en el 4,4% de los pacientes. En el estudio SENSCIS realizado en pacientes con SSc-ILD, se informó diarrea en el 75,7% versus el 31,6% de los pacientes tratados con OFEV® y placebo, respectivamente. La diarrea condujo a una reducción de la dosis de OFEV® en el 22,2% de los pacientes y a la interrupción de OFEV® en el 6,9% de los pacientes (véase la sección Reacciones adversas). La diarrea debe tratarse ante la aparición de los primeros síntomas con hidratación adecuada y con medicamentos antidiarreicos, p. ej., loperamida, y puede requerir la interrupción del tratamiento. El tratamiento con OFEV® podrá reanudarse en una dosis reducida (100 mg dos veces al día) o en la dosis completa (150 mg dos veces al día). En el caso de que persista un cuadro de diarrea grave a pesar del tratamiento sintomático, deberá interrumpirse el tratamiento con OFEV®. Náusea y vómito: Las náuseas y los vómitos fueron eventos adversos informados con frecuencia (véase la sección Reacciones Adversas). En la mayoría de los pacientes con náuseas y vómitos, el evento fue de intensidad leve a moderada. En los estudios INPULSIS, las náuseas condujeron a la interrupción del tratamiento con OFEV® en el 2,0% de los pacientes y los vómitos condujeron a la interrupción de este fármaco en el 0,8% de los pacientes. En el estudio SENSCIS, la frecuencia de náuseas y vómitos que causaron la interrupción del tratamiento con OFEV® fue del 2,1% y 1,4%, respectivamente. Si los síntomas persisten a pesar de haberse instaurado un tratamiento de soporte adecuado (lo que incluye tratamiento antiemético), puede ser necesario implementar una reducción de la dosis o la interrupción del tratamiento. El tratamiento podrá reanudarse en una dosis reducida (100 mg dos veces al día) o en la dosis completa (150 mg dos veces al día). Ante la presencia de síntomas severos que persistan, deberá interrumpirse el tratamiento con OFEV®. La diarrea y los vómitos pueden provocar deshidratación con o sin alteraciones electrolíticas, lo que podría conducir a un deterioro de la función renal. Función hepática: La seguridad y la eficacia de OFEV® no han sido estudiadas en pacientes con insuficiencia hepática moderada (Child Pugh B) o severa (Child Pugh C). Por lo tanto, no se recomienda el tratamiento con OFEV® en dichos pacientes. Sobre la base de que existe una mayor exposición, es posible que los pacientes con insuficiencia hepática leve (Child Pugh A) corran más riesgos de sufrir eventos adversos. Los pacientes con insuficiencia hepática leve (Child Pugh A) deben tratarse con una dosis reducida de OFEV® (ver las secciones Dosis y vía de administración, Farmacocinética.) Se han observado casos de la lesión hepática producida por medicamentos con el tratamiento con nintedaib. En el periodo posterior a la comercialización, se han informado casos serios y casos no serios de daño hepático causado por el medicamento, incluso daño hepático grave con desenlace mortal. La mayoría de los eventos hepáticos ocurren dentro de los primeros tres meses de tratamiento. Por lo tanto, deben determinarse los niveles de bilirrubina y transaminasas hepáticas antes de iniciarse el tratamiento con OFEV®, a intervalos periódicos durante los primeros tres meses de tratamiento y luego a intervalos periódicos (p. ej., en cada visita del paciente) o según esté clínicamente indicado. Las elevaciones de las enzimas hepáticas (ALT, AST, ALKP, gamma glutamiltransferasa (GGT)) y de los valores de bilirrubina fueron reversibles con la reducción de la dosis o la interrupción del tratamiento, en la mayoría de los casos. En el caso de detectarse elevaciones de las transaminasas (AST o ALT) > 3 veces el límite normal superior (ULN), se recomienda la reducción de la dosis o la interrupción del tratamiento con OFEV® y el monitoreo estrecho del paciente. Una vez que las transaminasas hayan retornado a los valores basales, el tratamiento con OFEV® podrá incrementarse nuevamente a la dosis completa (150 mg dos veces al día) o bien reiniciarse en una dosis reducida (100 mg dos veces al día), que luego podrá incrementarse hasta llegar a la dosis completa (véase la sección Dosis y vía administración). Si alguna de estas elevaciones en los parámetros de la función hepática estuviera asociada con signos o síntomas clínicos de lesión hepática, p. ej., ictericia, deberá interrumpirse en forma definitiva el tratamiento con OFEV®. Deben investigarse las posibles causas alternativas de las elevaciones de las enzimas hepáticas. Los pacientes con bajo peso corporal ( < 65 kg), los de raza asiática y las mujeres tienen un mayor riesgo de elevaciones de las enzimas hepáticas. La exposición a nintedanib se incrementó de manera lineal en función de la edad de los pacientes, lo que también puede dar lugar a un mayor riesgo de desarrollar elevaciones de las enzimas hepáticas (véase la sección Farmacocinética). Se recomienda un monitoreo estrecho en los pacientes que presenten estos factores de riesgo. Hemorragia: La inhibición del VEGFR podría estar asociada con un mayor riesgo de sangrado. En los estudios con OFEV®, la frecuencia de pacientes que tuvieron eventos adversos de sangrado fue ligeramente más alta en el grupo tratado con OFEV® (10,3%) para INPULSIS; 11,1% para SENSCIS) que en el grupo tratado con placebo (7,8% para INPULSIS; 8,3% para SENSCIS). La epistaxis no seria fue el evento de sangrado más frecuente. En los estudios INPULSIS los eventos de sangrado serios se produjeron con frecuencias bajas y similares en los 2 grupos de tratamiento (placebo: 1,4%; OFEV®: 1,3%). En el estudio SENSCIS, los eventos de sangrado serios se produjeron con frecuencias bajas en ambos grupos de tratamiento (placebo: 0,7%; OFEV®: 1,4%). Los pacientes que tenían un riesgo conocido de sangrado, lo que incluye a los pacientes con una predisposición hereditaria al sangrado o los pacientes que estaban recibiendo una dosis completa de tratamiento anticoagulante, no fueron incluidos en los estudios clínicos. Por lo tanto, el tratamiento con OFEV® en estos pacientes podrá implementarse únicamente en el caso de que el beneficio previsto supere el potencial riesgo implicado. En el periodo posterior a la comercialización, se han observado episodios de hemorragia graves y no graves, algunos de los cuales fueron mortales. Eventos tromboembólicos arteriales: Los pacientes con antecedentes recientes de infarto de miocardio o accidente cerebrovascular fueron excluidos de los estudios clínicos. En los estudios INPULSIS, los eventos tromboembólicos arteriales fueron eventos infrecuentes: se informaron en el 0,7% de los pacientes en el grupo tratado con placebo y en el 2,5% de los pacientes en el grupo de tratamiento con OFEV®. Mientras que los eventos adversos que reflejan una cardiopatía i