OHRENCIA®
BRISTOL M.S.
Denominación genérica: Abatacept.
Forma farmacéutica y formulación: Cada frasco ámpula con solución contiene (Vía de administración: intravenosa): Abatacept 250 mg. Excipiente cbp. Cada jeringa prellenada con solución contiene (Vía de administración: subcutáneo): Abatacept 125 mg. Excipiente cbp. Proteína de fusión de origen de ADN recombinante expresado en células de ovario de hámster chino (CHO).
Descripción: OHRENCIA® (abatacept), un modulador selectivo de la co-estimulación, es una proteína de fusión soluble que consiste en el dominio extracelular del antígeno 4 asociado con linfocitos T citotoìxico humano (CTLA-4) enlazado a la porción Fc modificada (dominios bisagra, CH2, y CH3) de la inmunoglobulina humana G1. Abatacept es producido mediante tecnología de ADN recombinante en un sistema de expresión de células de mamífero. El peso molecular aparente de abatacept es de 92 kilodaltones. OHRENCIA® INTRAVENOSO: se suministra como un polvo estéril, blanco, libre de conservadores, liofilizado para administración intravenosa. Después de la reconstitución con 10 mL de agua estéril para inyección, la solución de OHRENCIA® es clara, de incolora a amarillo pálido, con un rango de pH de7.2 a 7.8. OHRENCIA® Subcutáneo: se suministra como una solución Inyectable estéril, libre de conservadores, lista para usarse en inyección subcutánea. El producto farmacéutico para inyección subcutánea es suministrado en una jeringa de vidrio prellenada desechable de dosis única con o sin guarda aguja La jeringa de vidrio Tipo I tiene un tapón recubierto y una aguja fija de acero inoxidable (bisel 5, calibre 29 de pared delgada, aguja de 1/2 pulgada) protegida con una cubierta de aguja rígida. Un exceso suficiente de abatacept es incorporado en cada jeringa para compensar las pérdidas aguja-jeringa, de tal forma que pueda entregarse 1.0 mL de solución con 125 mg de abatacept durante la administración subcutánea. La solución subcutánea es clara, incolora a amarilla pálida con un pH de 6.8 a 7.4.
Indicaciones terapéuticas: Artritis reumatoide (ar) en adultos: OHRENCIA® Puede ser administrado como infusión intravenosa (IV) o por inyección subcutánea (SC) para artritis reumatoide (AR) en adultos: OHRENCIA® está indicado para reducir signos y síntomas, inducir una respuesta clínica mayor, inhibir la progresión de daño estructural, y mejorar la función física en pacientes adultos con artritis reumatoide activa de moderada a grave. OHRENCIA® puede ser usada como monoterapia o en combinación con otros medicamentos anti-reumáticos modificadores de la enfermedad (DMARD, por sus siglas en inglés) diferentes a agentes antagonistas de factor de necrosis tumoral (TNF). Artritis juvenil idiopática: OHRENCIA® debe ser administrado como infusión intravenosa (IV) para Artritis idiopática juvenil OHRENCIA® IV está indicado para reducir signos y síntomas en pacientes pediátricos de 6 años y mayores con artritis idiopática juvenil poliarticular con actividad de moderada a grave. OHRENCIA® puede ser usado como monoterapia o en combinación con metotrexato (MTX). Limitaciones importantes para su uso: OHRENCIA® no debería ser usado en combinación con antagonistas del Factor de Necrosis Tumoral (TNF, por sus siglas en inglés), (Ver Precauciones generales: Combinación con agentes antagonistas del TNF). OHRENCIA® no está recomendado para usarse en combinación con otros medicamentos biológicos para la artritis reumatoide como anakinra (Ver Indicaciones medicamentosas y de otro género).
Farmacocinética y farmacodinamia: Farmacocinética: Adultos Sanos y adultos con AR: Absorción: Abatacept se administra por vía intravenosa. Distribución: La farmacocinética de abatacept fue estudiada en adultos sanos después de una infusión intravenosa única de 10mg/kg y en pacientes con AR después de infusiones intravenosas múltiples de 10mg/kg (ver Tabla 1).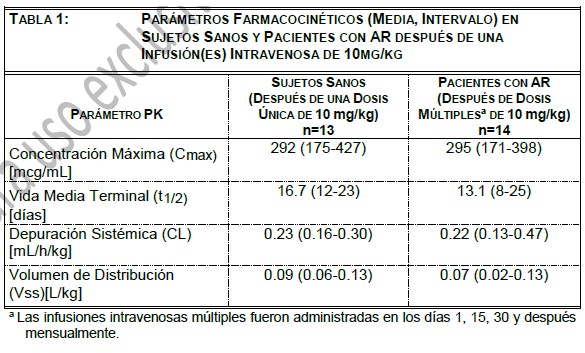
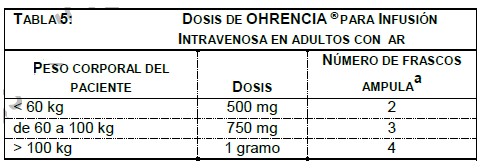
La farmacocinética de abatacept en pacientes con AR y sujetos sanos pareció ser comparable. En pacientes con AR, después de infusiones intravenosas múltiples, la farmacocinética de abatacept mostró incrementos proporcionales de Cmax y ABC sobre el intervalo de dosis de 2 mg/kg a 10mg/kg. A dosis de 10mg/kg, la concentración sérica pareció alcanzar un estado estable en el día 60 con una concentración valle promedio (intervalo) de 24 (1-66) mcg/mL. No ocurrió ninguna acumulación sistémica de abatacept con un tratamiento continuo repetido con 10mg/kg en intervalos mensuales en pacientes con AR. Análisis farmacocinéticos poblacionales revelaron que se observó una tendencia hacia una depuración mayor de abatacept al incrementar el peso corporal. La edad y el género (cuando se corrigió el peso corporal) no afectó la depuración. El uso concomitante con MTX, AINEs, corticosteroides, y agentes antagonistas del TNF no influyeron en la depuración de abatacept. AR en Adulto - Régimen de dosificación: Subcutáneo: Abatacept mostró una farmacocinética lineal después de la administración subcutánea. La media (rango) de Cmin y Cmax en el estado estacionario observadas después de 85 días de tratamiento fueron de 32.5 mcg/mL (6.6 a 113.8 mcg/mL) y 48.1 mcg/mL (9.8 a 132.4 mcg/mL), respectivamente. La biodisponibilidad de abatacept después de la administración subcutánea en relación con la administración intravenosa es de 78.6%. La media del estimado de la depuración sistémica (0.28 mL/h/kg), volumen de distribución (0.11 L/kg), y vida media terminal (14.3 días) fueron comparables entre la administración SC e IV. Se realizó un estudio único para determinar el efecto del uso de la monoterapia con abatacept en la inmunogenicidad después de la administración subcutánea sin una carga IV. Cuando la dosis de carga IV no fue administrada, se alcanzó una media de la concentración mínima de 12.6 mcg/mL después de 2 semanas de dosificación. La respuesta de eficacia a través del tiempo en este estudio parece ser consistente con los estudios que incluyeron una dosis de carga IV, sin embargo, el efecto de no administrar la dosis de carga IV en el establecimiento de la eficacia no fue estudiado formalmente. De acuerdo al análisis de farmacocinética en la población de pacientes con AR en donde abatacept fue administrado de manera subcutánea, la depuración de abatacept tendió a ser mayor con el incremento en el peso corporal, lo cual fue consistente con los datos de I.V. La edad y el género (corregidos para el peso corporal) no afectaron la depuración aparente. MTX, NSAID, corticoesteroides, y agentes bloqueadores del TNF concomitantes no influyeron en la depuración aparente de abatacept. Metabolismo y eliminación: No se llevaron a cabo estudios para evaluar el metabolismo o la eliminación de abatacept en humanos. Debido a consideraciones estéricas e hidrofílicas, el abatacept no se metabolizaría por las enzimas hepáticas del citocromo P450. Debido al gran peso molecular no se espera que abatacept se elimine por vía. Artritis idiopática juvenil: El análisis farmacocinético poblacional de los datos de concentraciones séricas en pacientes con AIJ de 6 a 17 años después de la administración de abatacept 10 mg/kg mostraron que la depuración estimada de abatacept cuando se normalizó por el peso corporal inicial fue más alto en pacientes con AIJ (0.44mL/h/kg) que en pacientes con AR. Después de considerar el efecto del peso corporal, la depuración de abatacept no se relacionó con la edad ni el género. Los estimados promedios del volumen de distribución y la vida media de eliminación fueron de 0.12 l/kg y 11.2 días, respectivamente. Como resultado de mayor depuración normalizada por peso corporal en los pacientes con AIJ, la exposición sistémica prevista de abatacept fue menor que la observada en adultos, de manera tal que las concentraciones promedio (intervalo) pico y valle observadas fueron 217 (57 a 700) y 11.9 (0.15 a 44.6) mcg/mL, respectivamente. El uso concomitante de metotrexato, corticoesteroides y AINEs tampoco mostro influencia en la depuración de abatacept en pacientes con AIJ Poblaciones especiales: Las características farmacocinéticas de abatacept no han sido estudiadas en niños ni adolescentes. No se efectuaron estudios formales para examinar el efecto de daño renal o daño hepático sobre la farmacocinética de abatacept. Farmacodinamia: Los estudios para determinar la dosis se llevaron a cabo con monoterapia con abatacept (placebo, 0.5 mg/kg, 2 mg/kg, y 10 mg/kg) y en combinación con MTX (placebo, 2 mg/kg, y 10 mg/kg). En ambos estudios, la tasa de respuesta del American College of Rheumatology (ACR) 20 se elevó al incrementar la dosis de 2 mg/kg a 10 mg/kg. En estudios clínicos con OHRENCIA®, utilizando dosis de aproximadamente 10 mg/kg, se observó inhibición de la activación de linfocitos T, disminución de productos de macrófagos, sinoviocitos de tipo fibroblastos, y células B, y reducciones en reactantes de inflamación de fase aguda. Se observó disminución en: niveles séricos de receptor para interleucina-2 soluble, un marcador de activación de linfocitos T; interleucina-6 sérica, un producto de macrófagos activados y sinoviocitos de tipo fibroblastos; factor reumatoide, un anticuerpo producido por células plasmáticas; y proteína C-reactiva, un reactante de inflamación de fase aguda. Además, bajaron los niveles séricos de metaloproteinasa-3 de matriz que produce la destrucción de cartílago y remodelación tisular. Se observaron también reducciones en el TNFa sérico. Estos cambios son consistentes con el mecanismo de acción de este modulador selectivo de la co-estimulación. Mecanismo de acción: Abatacept modula selectivamente una señal de co-estimulación clave requerida para una activación completa de linfocitos T que expresan CD28. Los linfocitos T se encuentran en el sinovio de pacientes con AR. Los linfocitos T activados contribuyen a la patogénesis de AR y otras enfermedades auto-inmunes. La activación completa de linfocitos T requiere de dos señales proporcionadas por células presentadoras de antígeno: reconocimiento de un antígeno específico por un receptor para células T (señal 1) y una segunda señal co- estimuladora. Una vía coestimuladora mayor incluye la unión de moléculas CD80 y CD86 en la superficie de células presentadoras de antígeno con el receptor CD28 en linfocitos T (señal 2). Abatacept se une específicamente a CD80 y CD86 inhibiendo selectivamente esta vía co-estimuladora. Estudios indican que las respuestas de linfocitos T naive son más afectadas por abatacept que las respuestas de linfocitos T de memoria. Estudios in vitro y en modelos animales demuestran que abatacept atenúa las respuestas de anticuerpos dependientes de linfocitos T e inflamación. In vitro, abatacept atenúa la activación de linfocitos T de conformidad con lo medido por una disminución de la proliferación y producción de citocinas en linfocitos humanos. Abatacept disminuye la producción antígeno-específica de TNFa, interferón-c, e interleucina-2 por linfocitos T. En un modelo de artritis inducida por colágena de rata, abatacept suprime la inflamación, disminuye la producción de anticuerpos anti- colágena y reduce la producción del antígeno específico de interferón-c. Información de eficacia de estudios clínicos: estudios clínicos en adultos con artritis reumatoide: La eficacia y seguridad de OHRENCIA® fueron evaluadas en seis estudios aleatorizados, doble ciego, controlados con placebo en pacientes ≥ 18 años con diagnóstico de AR activa de acuerdo con los criterios de American College of Rheumatology (ACR). Los estudios están designados de la siguiente manera: Estudio I (IM103002), Estudio II (IM101100), Estudio III (IM101102 AIM), Estudio IV (IM101029, ATTAIN), Estudio V (IM101031, ASSURE) y Estudio VI (IM101023, AGREE). Los estudios I, II, III, IV Y VI requirieron pacientes que tuvieran al menos 12 articulaciones sensibles a la presión y 10 inflamadas en la aleatorización. El estudio V no requirió ningún número específico de articulaciones sensibles a la presión o inflamadas. Se administró OHRENCIA® o placebo en forma de una infusión intravenosa en la semana 0, 2 y 4 y después una vez al mes. El Estudio SC-I fue un estudio de no inferioridad, doble simulación, doble ciego, aleatorizado que comparó la eficacia y seguridad de abatacept administrado por vía subcutánea después de una dosis única de abatacept por vía intravenosa en sujetos con artritis reumatoide (AR), que recibían metotrexato de base (MTX), y que experimentaron una respuesta inadecuada a MTX (MTX-IR). Para ensayos clínicos adicionales en adultos con artritis reumatoide, vea la sección de "Respuesta Clínica". En el Estudio I se evaluó OHRENCIA® como monoterapia en 122 pacientes con AR activa los cuales habían fallado con al menos un DMARD no biológico o etanercept. En el Estudio II y Estudio III, se determinó la eficacia y seguridad de OHRENCIA® en pacientes con una respuesta inadecuada a MTX y que continuaron con su dosis estable de MTX. En el Estudio IV, se determinó la eficacia y seguridad de OHRENCIA® en pacientes con una respuesta inadecuada a un agente antagonista del TNF, con el agente antagonista del TNF suspendido antes de la aleatorización; otros DMARDs fueron permitidos. El Estudio V principalmente evaluó la seguridad en pacientes con AR activa que requieren de intervención adicional a pesar de la terapia actual con DMARDs; todos los DMARDs utilizados en el reclutamiento se continuaron. En el Estudio VI, la eficacia y seguridad de OHRENCIA® fueron evaluadas en pacientes vírgenes al MTX con AR temprana, erosiva (≤2 años de duración de la enfermedad). En el Estudio VI, pacientes previamente vírgenes a MTX fueron aleatorizados para recibir OHRENCIA® más MTX o MTX más placebo. En el Estudio SC-I, el objetivo fue demostrar la no inferioridad de la eficacia y seguridad de OHRENCIA® subcutáneo comparado con la administración intravenosa en sujetos con AR activa de moderada a grave y que experimentaran una respuesta inadecuada a MTX. En el estudio VI, la seguridad y eficacia de abatacept fue evaluada en pacientes sin experiencia previa a MTX, con factor reumatoide (FR) y/o al péptido 2 anticíclico citrulinado (anti-CCP2) positivo, con artritis reumatoide erosiva temprana ( < 2 años de duración de la enfermedad), los cuales fueron fueron aleatorizados al recibir OHRENCIA® más MTX o MTX más placebo El Estudio I los pacientes fueron aleatorizados para recibir una de tres dosis de OHRENCIA® (0.5, 2 ó 10 mg/kg) o placebo finalizando en la semana 8. En el Estudio II los pacientes fueron aleatorizados para recibir 2 ó 10 mg/kg de OHRENCIA® o placebo por 12 meses. Para los Estudios I y II, solo los resultados en el grupo de 10mg/kg se discuten abajo. En el Estudio III, IV, V y VI, los pacientes fueron aleatorizados para recibir una dosis fija de aproximadamente 10mg/kg de OHRENCIA® o placebo durante 12 meses (Estudios III, V y VI) ó 6 meses (Estudio IV). La dosis de OHRENCIA® fue de 500 mg para pacientes que pesan menos de 60 kg, 750 mg para pacientes que pesen de 60 a 100 kg, y 1 g para pacientes que pesan más de 100 kg. En el Estudio SC-I, OHRENCIA® fue administrado por vía subcutánea a los pacientes estratificados por peso corporal ( < 60 kg, 60 a 100 kg, > 100 kg) después de una dosis única de carga de OHRENCIA® intravenoso y posteriormente cada semana. Los sujetos continuaron tomando su dosis actual de MTX desde el día de la aleatorización. Respuesta clínica: Respuesta ACR: El porcentaje de pacientes tratados con OHRENCIA® que alcanzaron respuestas ACR 20, 50, y 70 y respuesta clínica mayor (definida al alcanzar una respuesta ACR 70 por un periodo continuo de 6 meses) en los Estudios I, III, IV y VI se muestran en la TABLA 2. Las tasas de respuestas a los 6 y 12 meses en el Estudio II para el grupo de 10mg/kg fue similar al grupo de OHRENCIA® en el Estudio III. En los Estudios III y IV, se observó mejoría en las tasas de respuesta ACR 20 en comparación con placebo en la evaluación del día 15, después de la administración de la primera dosis y se mantuvo durante el periodo doble ciego del estudio. En el estudio VI, la mejoría en la tasa de respuesta ACR 50 en pacientes tratados con OHRENCIA® más MTX en comparación de los pacientes tratados con MTX más placebo se observó a los 29 días y se mantuvo durante el trascurso del estudio doble ciego. La respuesta ACR 50 con OHRENCIA® fue significativamente mayor que con placebo en los meses 2 y 3, respectivamente, para los Estudios III y IV, con mejora continua en la tasa de respuesta ACR 20 durante el periodo doble ciego (mes 12 en el Estudio III y mes 6 en el Estudio IV). En los periodos controlados con placebo en los Estudios II, III y VI, las tasas de respuesta de ACR se mantuvieron por 12 meses en los pacientes tratados con OHRENCIA®. En la extensión abierta no controlada, a largo plazo de los Estudios II, III, IV y VI, se observaron respuestas duraderas y sostenidas de ACR 20, 50 y 70 durante 7, 5, 5, y 2 años respectivamente en base a los análisis observados. En el estudio II, la respuesta ACR fue evaluada a los 7 años con 31/43 (72%) respuesta de ACR 20, 25/43 (58%) respuesta ACR 50, y 19/43 (44%) respuesta ACR 70. En el estudio III, la respuesta ACR fue evaluada a los 5 años con 224/268 (84%) respuesta ACR 20, 165/20 (61%) de respuesta ACR 50, y 107/270 (40%) respuesta ACR 70. En el estudio IV, la respuesta ACR fue evaluada a los 5 años con 66/89 (74%) respuesta ACR 20, 45/88 (51%) respuesta ACR 50, y 21/91 (23%) respuesta ACR 70. En el estudio VI, la respuesta ACR fue evaluada a los 2 años con 196/219 (90%) de respuesta ACR 20, 169/217 (78%) de respuesta ACR 50, y 124/216 (57%) de respuesta ACR 70. Se observó una mayor mejoría en todos los componentes de criterio de la respuesta ACR en los pacientes tratados con OHRENCIA® que en los pacientes tratados con placebo a los 6 (Estudio IV) y 12 meses (Estudio II y III). En el estudio VI, se observó una mayor mejoría en todos los componentes ACR a los 12 meses en pacientes tratados con OHRENCIA® más MTX que en aquellos pacientes tratados con MTX más placebo. En la extensión abierta de los Estudios II, III y IV, se mantuvieron las mejoras en los componentes ACR individuales a los 7, 5 y 5 años, respectivamente, del tratamiento con OHRENCIA®. En el Estudio SC-I, OHRENCIA® administrado por vía subcutánea (SC) no fue inferior a las infusiones (IV) de OHRENCIA® con respecto a las respuestas ACR 20 durante 6 meses de tratamiento. Los pacientes tratados con OHRENCIA® SC también lograron respuestas ACR 50 y 70 similares a las de los pacientes que recibieron OHRENCIA® intravenosamente a 6 meses. No se observó diferencia en la respuesta clínica entre los 3 grupos de peso.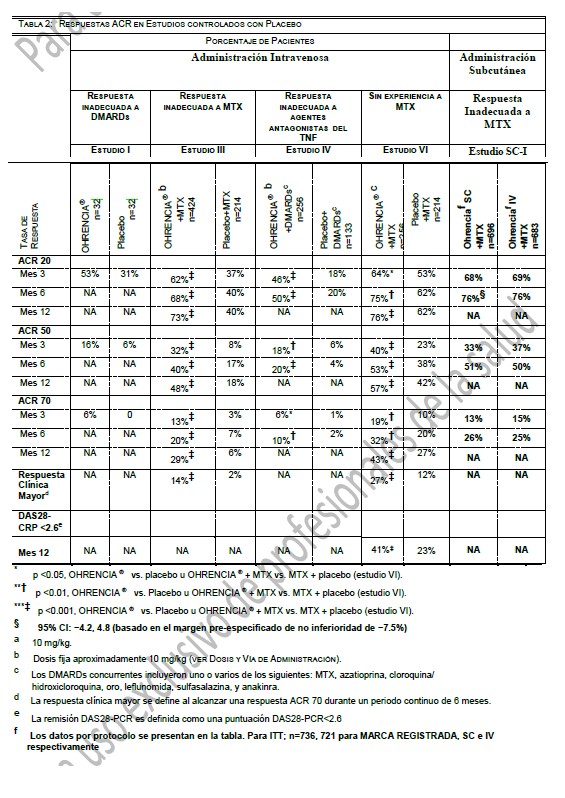
Entre los pacientes tratados con OHRENCIA® en el estudio III, 14% alcanzaron una respuesta clínica mayor, en comparación con 2% de los pacientes que recibieron placebo. Además, 6% de pacientes tratados con OHRENCIA® en este estudio de 12 meses alcanzaron una respuesta clínica mayor extendida (respuesta ACR 70 continua durante 9 meses) en comparación con 0.5% en pacientes que recibieron placebo. En el Estudio III, para los pacientes tratados con OHRENCIA® por mas de 2 años incluyendo periodos doble ciegos y abiertos, el porcentaje de sujetos que alcanzaron una respuesta clínica mayor y una respuesta clínica mayor extendida se incrementó a 34.3% y 24.5%, respectivamente. Los pacientes tratados con OHRENCIA® experimentaron una mayor mejora en la rigidez matutina que los pacientes que recibieron placebo. Respuesta das28: La actividad de la enfermedad fue también evaluada empleando el Resultados de Actividad de Enfermedad 28 (DAS28 VSG). En los Estudios III y IV, el DAS28 promedio del nivel basal fue de 6.8 y 6.9 unidades, respectivamente, representando un grado mayor de actividad de la enfermedad. En el Estudio III, la mejora promedio en DAS28 en el mes 12 en los pacientes tratados con OHRENCIA® de 2.9 fue significativamente mayor que la mejora promedio de 1.5 observada en pacientes que recibieron placebo. La remisión definida por DAS28 se alcanzó en el 17% de los pacientes tratados con OHRENCIA® en comparación con el 2% de los pacientes que recibieron placebo a los 12 meses. En el Estudio IV, en el mes 6, se observó una mejora significativa mayor en DAS28 en los pacientes tratados con OHRENCIA® que en los pacientes que recibieron placebo (reducción de 2.0 vs 0.7 unidades, respectivamente). La remisión definida por DAS28 se alcanzó en 10% de los pacientes tratados con OHRENCIA® en comparación con1% de los pacientes que recibieron placebo a los 6 meses. En el Estudio VI, los pacientes tratados con OHRENCIA® más MTX tuvieron una alta tasa de remisión de DAS28- PCR a los 12 meses que en aquellos tratados con MTX más placebo (Tabla 2). De los pacientes tratados con OHRENCIA® mas MTX quienes alcanzaron una remisión de DAS28-PCR, 54% no tenían articulaciones activas, 17% tenía alguna articulación activa, 7% tenían dos articulaciones activas, y 22% tenían tres o más articulaciones activas, se consideró como una articulación activa cuando se clasificó como dolorosa, inflamada, o ambas. Respuesta radiográfica: Se evaluó radiográficamente el daño estructural a las articulaciones en un periodo de 2 años en el Estudio III. Los resultados fueron medidos utilizando La escala total de Sharp modificada por Genant (TSS) y sus componentes, la escala de erosión y la escala de Disminución del Espacio Articular (JSN). El TSS promedio a nivel basal fue de 31.7 en los pacientes tratados con OHRENCIA® y 33.4 en pacientes que recibieron placebo. En el primer año, los pacientes recibieron OHRENCIA® o placebo de modo doble ciego. OHRENCIA® /MTX inhibieron la progresión del daño estructural en comparación con placebo/MTX después de 12 meses de tratamiento así como se observa en la TABLA 3. La inhibición de la progresión del daño estructural con OHRENCIA® fue observada independientemente de la duración de la enfermedad (menos de 2 años, 2 a 5 años, 5 a 10 años, y más de 10 años)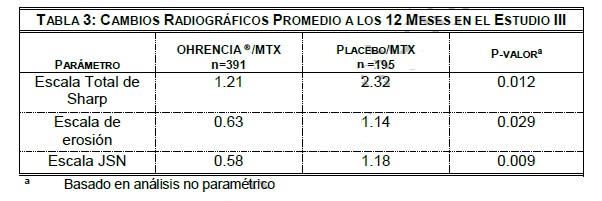
En la extensión abierta del Estudio III, el 75% (n=324) de los pacientes aleatorizados inicialmente a OHRENCIA® /MTX fueron evaluados radiográficamente por TSS. Después de 2 años de tratamiento con OHRENCIA® /MTX, se observó la inhibición de la progresión del daño estructural. El 50% de los pacientes no tuvieron progresión del daño estructural definido como un cambio en el TSS de cero o menos en 2 años. El 86% de los pacientes sin progresión radiográfica después de 1 año de tratamiento con OHRENCIA® /MTX, no tuvo progresión en 2 años. Para los pacientes tratados con OHRENCIA® /MTX, el cambio promedio en TSS del año 1 al año 2 fue 57% menor que el cambio promedio en TSS de la línea basal al año 1. Con base a las evaluaciones anuales, se observó una disminución de la progresión radiográfica en las 3 escalas, el mayor decremento fue observado en el primer año de tratamiento con abatacept en los periodos no controlados, abiertos, a largo plazo. Al final del periodo a largo plazo (4 años, 1821 días), 106/235 (45.1%) de los sujetos en el grupo original de abatacept y 45/115 (39.1%) de los sujetos en grupo original de placebo no mostraron progresión radiográfica basada en los resultados totales. En el estudio VI, el cambio promedio en el TSS a los 12 meses fue poco significativo en pacientes tratados con OHRENCIA® más MTX comparado con aquellos pacientes tratados con MTX más placebo. A los 12 meses 61% (148/242) de los pacientes tratados con abatacept mas metotrexato y 53% (128%242) de los pacientes tratados con metotrexato más placebo no tuvieron progresión (cambio desde el nivel basal en TSS≤0). Entre los pacientes incluidos en el periodo de 12 meses de estudio abierto, la progresión del daño estructural fue menor en aquellos que recibieron abatacept más metotrexato continuamente (por 24 meses) en comparación con los pacientes que recibieron inicialmente metotrexato mas placebo (12 meses) y fueron cambiados a abatacept mas metotrexato por los siguientes 12 meses. De estos pacientes, 57% (121/213) quienes recibieron abatacept más metotrexato de forma continua y 44% (84/192) de los pacientes que recibieron inicialmente metotrexato y cambiaron a la combinación con abatacept, no mostraron progresión. Respuesta de la función física: La mejora de la función física fue medida por el Índice de Discapacidad del Cuestionario de Evaluación de Salud (HAQ-DI) en los Estudios III, IV, V y VI y un HAQ-DI modificado en el Estudio II. En los estudios II-V, OHRENCIA® demostró una mejora significativamente mayor a partir de la evaluación basal que placebo en HAQ-DI, y una proporción significativamente mayor de pacientes tratados con OHRENCIA® en comparación con placebo mostraron una mejora clínicamente significativa (reducción en HAQ-DI de ≥0.3 unidades a partir de la evaluación basal). En el estudio VI, se observó una mejora significativamente mayor a partir de la evaluación basal en HAQ-DI en pacientes tratados con OHRENCIA® más MTX comparado con pacientes tratados con MTX más placebo, y significativamente mas pacientes del grupo OHRENCIA® más MTX comparado con los del grupo MTX más placebo alcanzaron una mejora clínica significativa a los 12 meses. En el Estudio SC-I, la mejoría evaluada por HAQ-DI a los 6 meses y a través del tiempo con respecto a la basal fue similar entre la administración subcutánea e intravenosa. En el Estudio III, entre los que respondieron a HAQ en el mes 12, 88% mantuvo la respuesta al mes 18, y85% mantuvo la respuesta al mes 24. Los resultados de los Estudios II-IV se muestran en la Tabla 4. Durante el período abierto del los Estudios II, III, IV y VI, la mejora de la función física se mantuvo durante 7 años, 5 años, 5 años, 2 años, respectivamente.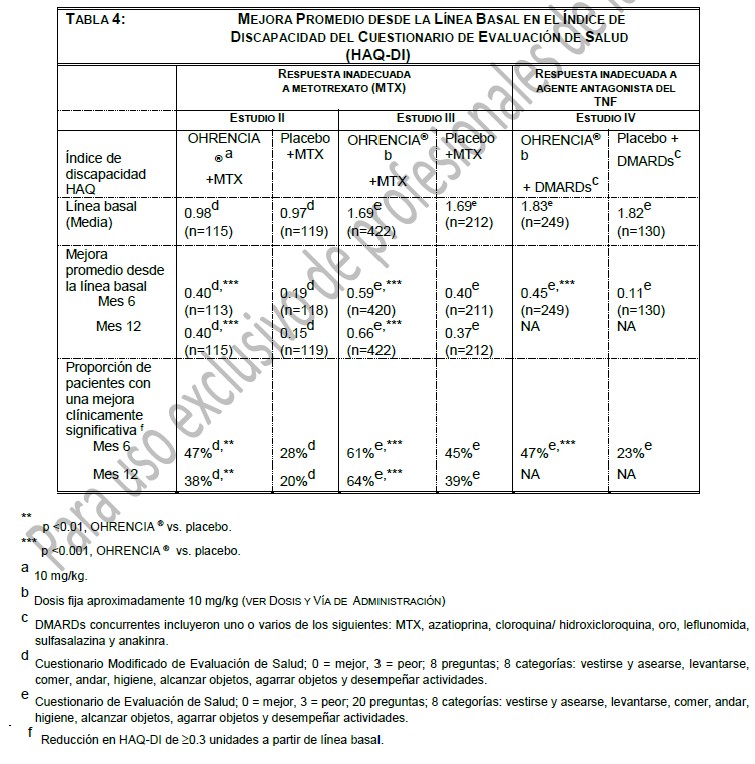
Resultados relacionados con la salud y calidad de vida: La calidad de vida relacionada con la salud fue evaluada mediante cuestionario SF-36 a los 6 meses en los Estudios II, III y IV y a los 12 meses en los Estudios II y III. En estos estudios, se observó una mejora clínica y estadísticamente significativa en el grupo OHRENCIA® en comparación con el grupo placebo en los 8 dominios de SF-36 (4 dominios físicos: función física, papel funcional, dolor corporal, salud general; y 4 dominios mentales: vitalidad, función social, papel emocional, y salud mental), así como el Resumen de Componentes Físicos (RCF) y el Resumen de Componentes Mentales (RCM). En el Estudio VI, se observó una mejoría a los 12 meses en el grupo de OHRENCIA® más MTX en comparación con el grupo de MTX más placebo en ambos resúmenes RCF y RCM y se mantuvo hasta los 24 meses. En los Estudios III y IV, se midió la fatiga por una Escala Análoga Visual de Fatiga validada, y se evaluaron los problemas de sueño mediante Índice de Problemas de Sueño (SPI) del Estudio de Desenlaces Médicos en el Modulo de Sueño. A los 12 meses y 6 meses, en los Estudios III y Estudio IV, respectivamente, se observaron reducciones estadísticamente significativas de fatiga y problemas de sueño, en pacientes tratados con OHRENCIA® en comparación con pacientes tratados con placebo. En el Estudio VI, se observó una gran reducción en los niveles de fatiga a los 6 y 12 meses en los pacientes tratados con OHRENCIA® más MTX que en aquellos pacientes tratados con MTX más placebo. En el tratamiento abierto con OHRENCIA®, los resultados de mejoras relacionadas a la salud y la calidad de vida, se han mantenido hasta por más de 4 años. Ensayos clínicos adicionales en artritis reumatoide en adultos: Estudio VII: abatacept o infliximab versus placebo. Se realizó un estudio aleatorizado, doble ciego, para evaluar la seguridad y eficacia de abatacept o infliximab versus placebo en pacientes con una respuesta inadecuada al metotrexato (Estudio VII, Estudio IM101043). Los pacientes del Estudio VII recibieron la misma dosis fija de abatacept que en los Estudios III-VI o 3 mg/kg de infliximab o placebo durante 6 meses. El Estudio VII continuó por 6 meses más con los grupos de abatacept e infliximab únicamente. El desenlace primario fue el cambio promedio en actividad de la enfermedad en pacientes tratados con abatacept en comparación con pacientes tratados con placebo a los 6 meses con una evaluación subsecuente doble ciego de la seguridad y eficacia de abatacept e infliximab a los 12 meses. El número de pacientes aleatorizados fue de 156 a abatacept, 165 a infliximab, y 110 a placebo. En el Estudio VII, los cambios promedio del DAS28 desde el inicio a los meses 6 a 12 se muestran en la tabla 5, así como los porcentajes de pacientes que alcanzaron DAS28 definidos con baja actividad de la enfermedad y remisión. La mayor mejoría (p < 0.001) en DAS28 se observó con abatacept e infliximab en comparación con placebo a seis meses en la porción controlada con placebo del ensayo; los resultados entre los grupos de abatacept e infliximab fueron similares. Se observó una mayor mejoría a los 12 meses con abatacept. Las respuestas ACR en el Estudio VII fueron consistentes con la puntuación DAS28. El periodo de etiqueta abierta del Estudio VII proporcionó una evaluación de la habilidad de abatacept de mantener la eficacia para sujetos originalmente aleatorizados a abatacept y la respuesta de eficacia de aquéllos sujetos que fueron cambiados a abatacept después del tratamiento con infliximab. La reducción desde el inicio en la puntuación DAS28 al día 365 (3.06) se mantuvo hasta el día 729 (3.34) en aquéllos pacientes que continuaron con abatacept. En aquéllos pacientes que inicialmente recibieron infliximab y después cambiaron a abatacept, hubo una mejoría en la puntuación DAS28 promedio al día 729 (3.07) en relación con el día 365 (3.88).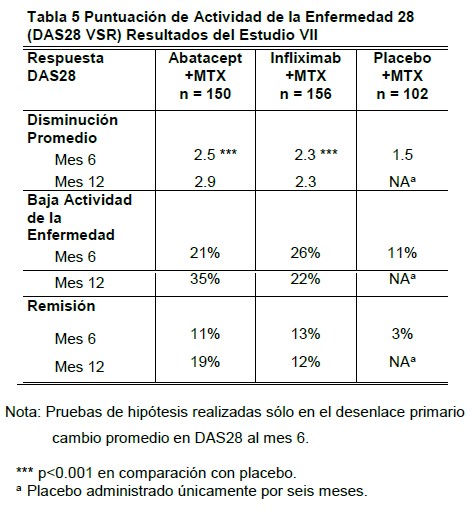
A los 6 meses, los eventos adversos graves considerados como relacionados con el tratamiento fueron del 1.9% (3 pacientes) en el grupo de abatacept, 4.8% (8) en el grupo de infliximab, y 2.7% (3) en el grupo de placebo. La frecuencia de infecciones graves fue del 1.3% (2) en el grupo de abatacept, 2.4% (4) en el grupo de infliximab, y 0.9% (1) en el grupo de placebo. La frecuencia de eventos adversos infusionales agudos fue del 5.1% (8) en el grupo de abatacept, 18.2% (30) en el grupo de infliximab, y 10.0% (11) en el grupo de placebo. A los 12 meses, los eventos adversos graves considerados en general como relacionados con el tratamiento fueron del 3.2% (5) en el grupo de abatacept y de 8.5% (14) en el grupo de infliximab. La frecuencia de infecciones graves fue del 1.3% (2) en el grupo de abatacept y 6.1% (10) en el grupo de infliximab, con un total de 5 infecciones oportunistas graves en el grupo de infliximab y ninguna en el grupo de abatacept. En relación con los valores anormales de laboratorio a los 6 meses, se desarrollaron anticuerpos antinucleares en el 1.7% (2) del grupo de abatacept, 32.2% (38) del grupo de infliximab, y 4.9% (4) del grupo de placebo. Estudio VIII: Seguridad de abatacept en pacientes con o sin periodo de lavado del tratamiento previo con bloqueadores del TNF: Se realizó un estudio de etiqueta abierta de abatacept con DMARDs no biológicos en pacientes con AR activa que tuvieron una respuesta inadecuada al tratamiento previo con bloqueadores del TNF (periodo de lavado de al menos 2 meses; n=449) o actual (sin periodo de lavado n=597) (Estudio VIII, Estudio IM101064). El desenlace primario, incidencia de eventos adversos, eventos adversos graves, y suspensiones por a eventos adversos durante 6 meses de tratamiento, fueron similares entre los pacientes que al momento de la inclusión eran usuarios previos y actuales de bloqueadores del TNF, asimismo la frecuencia de infecciones graves fue semejante en ambos grupos. Los resultados del Estudio VIII respaldan la transición del tratamiento con un agente bloqueador del TNF a tratamiento con OHRENCIA® en la próxima dosis programada del tratamiento con el agente bloqueador del TNF. Ensayos clínicos en artritis idiopática juvenil: La seguridad y eficacia de OHRENCIA® en niños con artritis idiopática Juvenil (AIJ) poliarticular fueron evaluadas en un estudio de tres partes (IM101033, AWAKEN) incluyendo una extensión de etiqueta abierta. Pacientes de 6 a 17 años de edad (n=190) con AIJ poliarticular con actividad moderada a grave que tuvieron una respuesta inadecuada a uno o más DMARDs, tales como MTX o bloqueadores del TNF, fueron tratados. Los pacientes tenían una duración de la enfermedad de aproximadamente 4 años con actividad moderada a grave al entrar al estudio, determinado mediante los conteos iniciales de articulaciones activas (promedio, 16) y articulaciones con pérdida de movilidad (promedio, 16); los pacientes tuvieron niveles elevados de proteína C reactiva (PCR) (promedio, 3.2 mg/dL) y VSG (promedio, 32 mm/h). Los pacientes incluidos tenían subtipos de AIJ que al inicio de la enfermedad incluían: Oligoarticular (16%), Poliarticular (64%; 20% fueron positivos al factor reumatoide), y Sistémico (20%). Al ingreso en el estudio, 74% de los pacientes estaban recibiendo MTX (dosis promedio, 13.2 mg/m2 por semana) y se mantuvieron con una dosis estable de MTX (aquéllos que no recibieron MTX no comenzaron tratamiento de MTX durante el estudio. En el Periodo A (etiqueta abierta, inducción), los pacientes recibieron 10 mg/kg (máximo de 1000 mg por dosis) por vía intravenosa en los días 1, 15, 29, y mensualmente de ahí en adelante. La respuesta fue evaluada utilizando la definición de mejoría ACR Pediátrica 30, definida como un ≥30% de mejoría en al menos 3 de las 6 variables principales de AIJ y ≥30% de empeoramiento en no más de 1 de las 6 variables principales de AIJ. Los pacientes que demostraron una respuesta ACR Pedi 30 al final del Periodo A fueron aleatorizados a la fase doble ciego (Periodo B) y recibieron OHRENCIA® o placebo durante 6 meses o hasta la reactivación de la enfermedad. Se definió reactivación de la enfermedad como un ≥30% de empeoramiento en al menos 3 de las 6 variables principales de AIJ con un ≥30% de mejoría en no más de 1 de las 6 variables principales de AIJ; ≥2 cm de empeoramiento de la Evaluación Global de Médicos o Padres fue necesaria si cualquiera era utilizada como 1 de las 3 variables principales de AIJ usadas para definir reactivación, y empeoramiento en ≥2 articulaciones fue necesario si el número de articulaciones activas o articulaciones con limitación de movimiento era usada como 1 de las 3 variables principales de AIJ utilizadas para definir reactivación. En la conclusión del Periodo A, las respuestas pediátricas ACR 30/50/70 fueron del 65%, 50%, y 28%, respectivamente. Las respuestas ACR 30 pediátricas fueron similares en todos los subtipos de AIJ estudiados. Durante la fase aleatorizada de retiro doble ciego (Periodo B), los pacientes tratados con OHRENCIA® experimentaron significativamente menos reactivación de la enfermedad en comparación con los pacientes tratados con placebo (20% vs 53%); IC 95% de la diferencia (15%, 52%). El riesgo de reactivación de la enfermedad entre los pacientes que continuaron con OHRENCIA® fue menor de un tercio que el riesgo para los pacientes retirados del tratamiento con OHRENCIA® (tasa de riesgo=0.31, IC 95% [0.16, 0.59]). Entre los pacientes que recibieron OHRENCIA® a lo largo del estudio (Periodo A, Periodo B, y el Periodo C de extensión de etiqueta abierta), la proporción de respondedores por ACR pediátrico 30/50/70 han permanecido consistente durante 31 meses.
Contraindicaciones: OHRENCIA® no debe administrarse a pacientes con hipersensibilidad conocida a OHRENCIA® o a cualquiera de sus componentes.
Precauciones generales: Combinación con agentes antagonistas del TNF: Existe poca experiencia con el uso de OHRENCIA® en combinación con agentes antagonistas del TNF (ver Farmacocinética y farmacodinamia en humanos: Estudios clínicos). En estudios clínicos controlados con placebo, en pacientes adultos con AR que reciben OHRENCIA® en combinación con terapia de agentes antagonistas del TNF, experimentaron más infecciones (24%) e infecciones graves (2.2%) comparado con pacientes tratados solo con agentes antagonistas del TNF (19% y 0.8%, respectivamente). No se recomienda la terapia concurrente con OHRENCIA® y un agente antagonista del TNF (Ver Interacciones medicamentosas y de otro género). Mientras se da la transición entre la terapia con agentes antagonistas del TNF y la terapia con OHRENCIA®, los pacientes deberán ser vigilados por si presentan signos de infección. Hipersensibilidad: Se pueden observar reacciones de hipersensibilidad durante el tratamiento con cualquier proteína inyectable. Tales reacciones han sido reportadas con la administración de OHRENCIA® en estudios clínicos, en donde no fue requerido que los pacientes recibieran tratamiento previo a la infusión previamente para evitar reacciones de hipersensibilidad. La aparición de anafilaxia fue rara en la experiencia con estudios clínicos abiertos, doble ciego, a largo plazo. La hipersensibilidad fue reportada de manera poco frecuente. Otros eventos posiblemente relacionados con hipersensibilidad a fármacos, como hipotensión, urticaria y disnea, que ocurrieron dentro de las 24 horas después de la infusión de OHRENCIA®, fueron poco comunes. (Ver Reacciones relacionadas con la infusión y reacciones de hipersensibilidad). Efectos sobre el sistema inmunológico: Existe la posibilidad que fármacos que afectan el sistema inmunológico, incluyendo OHRENCIA®, afecten las respuestas a vacunación y las defensas del huésped contra infecciones y enfermedades malignas. Infecciones: Se han reportado infecciones graves, incluyendo sepsis y neumonía, en pacientes que reciben OHRENCIA®. Algunas de estas infecciones han sido fatales. Muchas de las infecciones graves ocurrieron en pacientes con terapia inmunosupresora concomitante la cual, además de la enfermedad subyacente, pudo predisponerlos aun más a infecciones. Los médicos deben tomar precauciones al considerar el uso de OHRENCIA® en pacientes con historia de infección recurrente o condiciones subyacentes que pueden predisponerlos a infecciones o infecciones crónicas, latentes o localizadas. Los pacientes que desarrollan una nueva infección mientras están sometidos al tratamiento con OHRENCIA® deben ser vigilados estrechamente. La administración de OHRENCIA® debe ser suspendida si un paciente desarrolla una infección grave (ver Reacciones adversas: infecciones). Se ha observado una tasa mayor de infecciones graves en pacientes adultos con AR tratados concurrentemente con agentes antago