OPDIVO®
BRISTOL M.S.
Denominación genérica: Nivolumab.
Forma farmacéutica y formulación: Solución Inyectable. El frasco ámpula contiene: Nivolumab 100 mg. Vehículo cbp 10 mL. El frasco ámpula contiene: Nivolumab 40 mg. Vehículo cbp 4 mL
Descripción: Anticuerpo monoclonal humano IgG4 de origen ADN recombinante expresado en células de ovario de hámster chino (CHO). OPDIVO® es un líquido estéril, sin conservadores, no pirógeno, claro a opalescente, incoloro a amarillo pálido que puede contener partículas claras (pocas). OPDIVO® inyectable para infusión intravenosa se suministra en frascos ámpula de dosis única.
Indicaciones terapéuticas: Melanoma no resecable o metastásico: OPDIVO® está indicado para el tratamiento de pacientes con melanoma no resecable o metastásico. OPDIVO®, en combinación con ipilimumab, está indicado para el tratamiento de pacientes con melanoma no resecable o metastásico. Cáncer de pulmón de células no pequeñas: OPDIVO® está indicado para el tratamiento de pacientes con cáncer de pulmón de células no pequeñas (CPCNP) tanto de tipo escamoso como no escamoso metastásico que muestra progresión durante o después de la quimioterapia basada en platino. Los pacientes con alteraciones genéticas tumorales en EGFR o ALK deben haber experimentado progresión de la enfermedad con una terapia para estas alteraciones antes de recibir tratamiento con OPDIVO®. Cáncer de células renales: OPDIVO® está indicado para el tratamiento de pacientes con cáncer avanzado de células renales que han recibido terapia previa antiangiogénica incluidos los inhibidores de la tirosin cinasa. Linfoma de Hodgkin clásico: OPDIVO® está indicado en el tratamiento de pacientes con Linfoma de Hodgkin Clásico (cHL) que han recaído o progresado después de un trasplante autólogo de células progenitoras hematopoyéticas (HSCT), o también en aquellos que presentan falla posterior al uso de brentuximab vedotin post-trasplante. Cáncer de cabeza y cuello de tipo escamoso: OPDIVO® está indicado para el tratamiento de pacientes con cáncer de cabeza y cuello de tipo escamoso recurrente o metastásico (SCCHN) con progresión de la enfermedad o después de la terapia basada en platino.
Farmacocinética y farmacodinamia: Nivolumab es un anticuerpo monoclonal humano que bloquea la interacción entre PD-1 y sus ligandos, PD-L1 y PD-L2. Nivolumab es una inmunoglobulina IgG4 kappa con una masa molecular calculada en 146 kDa. Mecanismo de acción: La unión de los ligandos de PD-1, PD-L1 y PD-L2, con el receptor PD-1 que se encuentra en las células T, inhibe la proliferación de las células T y la producción de citocinas. El incremento de los ligandos de PD- 1 ocurre en algunos tumores y la señalización por esta vía puede contribuir a la inhibición de la vigilancia inmunitaria tumoral de las células T activas. Nivolumab es un anticuerpo monoclonal humano tipo inmunoglobulina G4 (IgG4) que se une con el receptor PD-1 y bloquea su interacción con PD-L1 y PD-L2, lo que libera la inhibición de la respuesta inmunitaria mediada por la vía PD-1, incluida la respuesta inmunitaria antitumoral. En modelos tumorales singénicos de ratón, el bloqueo de la actividad PD-1 redujo el crecimiento tumoral. La inhibición mediada de nivolumab (anti PD-1) e ipilimumab (anti CTLA-4) combinados, resulta en un incremento de la función de las células T, la cual es mayor que los efectos de cualquier anticuerpo solo, lo que resulta en una mejora en las respuestas antitumorales en melanoma metastásico. En modelos tumorales singénicos de murinos, el bloqueo dual de PD-1 y CTLA-4 dio como resultado un aumento en la actividad antitumoral. Farmacocinética: La farmacocinética de nivolumab se valoró con un enfoque de farmacocinética poblacional para OPDIVO® como agente único y OPDIVO® con ipilimumab. OPDIVO® como agente único: La farmacocinética de nivolumab se estudió en pacientes con un intervalo de dosis de 0.1 a 20 mg/kg administrados como dosis única o dosis múltiple de OPDIVO® cada 2 o 3 semanas. La depuración de nivolumab disminuye con el tiempo, con una reducción máxima media (% de coeficiente de variación [CV%]) desde los valores de referencia de aproximadamente 24.5% (47.6%) lo que da como resultado una separación geométrica del estadio estacionario medio (CLss) (CV%) de 8.2 mL/h (53.9%); la disminución en CLss no se considera clínicamente relevante. La media geométrica del volumen de distribución en estado estacionario (Vss) (CV%) es 6.8L (27.3%), y la media geométrica de la vida media de eliminación (t1/2) es 25 días (77.5%). Las concentraciones estables de nivolumab fueron alcanzadas aproximadamente a las 12 semanas cuando se administró a 3 mg/kg cada 2 semanas, y la acumulación sistémica fue aproximadamente 3.7 veces. La exposición a nivolumab aumentó la dosis de manera proporcional en el intervalo de dosis de 0.1 a 10 mg/kg con administración cada 2 semanas. OPDIVO® con ipilimumab: La media geométrica (CV%) de CL, Vss y de la vida media terminal de nivolumab fue de 10.0 mL/h (50.3%), 7.92 L (30.1%), y 24.8 días (94.3%), respectivamente. Cuando se administró en combinación, la CL de nivolumab se incrementó en un 24%, mientras que no se observó ningún efecto sobre la depuración de ipilimumab. Cuando se administra en combinación, la depuración de nivolumab se incrementó en un 42% en presencia de anticuerpos de anti-nivolumab. No hubo efecto de los anticuerpos anti-ipilimumab sobre la depuración de ipilimumab. Poblaciones específicas: Basado en un análisis de farmacocinética poblacional se presume que los siguientes factores no tienen importancia clínica en el efecto de la depuración de nivolumab: edad (29 a 87 años), peso (35 a 160kg), género, LDH basal, expresión de PD-L1, tipo de tumor, tamaño del tumor, daño renal y daño hepático leve. Insuficiencia renal: El efecto de la insuficiencia renal en la depuración de nivolumab se evaluó en un análisis de farmacocinética poblacional en pacientes con insuficiencia renal leve (eGFR 60 a 89 mL/min/1.73 m2; n=313), moderada (eGFR 30 a 59 mL/min/1.73 m2; n=140) o grave (eGFR 15 a 29 mL/min/1.73 m2; n=3). No se encontraron diferencias clínicamente importantes en la depuración de nivolumab entre pacientes con insuficiencia renal y pacientes con función renal normal. [Ver Precauciones generales, insuficiencia renal]. Insuficiencia hepática: El efecto de la insuficiencia hepática en la depuración de nivolumab se evaluó en un análisis de farmacocinética poblacional en pacientes con insuficiencia hepática leve (bilirrubina total [BT] menor o igual al límite superior normal [LSN] y AST mayor al LSN o BT menor de 1 a 1.5 veces el LSN y cualquier valor de AST; n=92). No se encontraron diferencias clínicamente importantes en la depuración de nivolumab entre pacientes con insuficiencia hepática leve, y pacientes con función hepática normal. Nivolumab no se ha estudiado en pacientes con insuficiencia hepática moderada (BT mayor de 1.5 a 3 veces el LSN y cualquier AST) o grave (BT mayor de 3 veces el LSN y cualquier AST). [Ver Precauciones generales, insuficiencia hepática].
Contraindicaciones: OPDIVO® no debe administrarse a pacientes con hipersensibilidad al biofármaco o a los componentes de la fórmula, embarazo, lactancia ni en menores de 18 años.
Precauciones generales: Los pacientes deben ser monitoreados continuamente (como mínimo hasta 5 meses después de la última dosis) ya que se puede producir una reacción adversa con nivolumab o con nivolumab en combinación con ipilimumab en cualquier momento, durante o después de la suspensión del tratamiento. Para sospechas de reacciones adversas mediadas inmunológicamente, se debe realizar una evaluación adecuada para confirmar esta etiología o excluir otra causa. De acuerdo a la gravedad de las reacciones adversas, se debe interrumpir el tratamiento con nivolumab o nivolumab en combinación con ipilimumab y se deben administrar corticosteroides. Si se emplea inmunosupresión con corticosteroides para tratar una reacción adversa, se debe iniciar una reducción progresiva de la dosis de al menos 1 mes de duración en cuanto se observe mejoría. Una disminución rápida de la dosis puede provocar un empeoramiento o recurrencia de la reacción adversa. Se debe añadir tratamiento inmunosupresor no corticosteroide si se observa un empeoramiento o no se produce una mejoría a pesar del uso de corticosteroides. Nivolumab o nivolumab en combinación con ipilimumab no se debe reanudar mientras el paciente esté recibiendo dosis inmunosupresoras de corticosteroides u otro tratamiento inmunosupresor. Se deben utilizar antibióticos profilácticos para prevenir la aparición de infecciones oportunistas en pacientes que reciben tratamiento inmunosupresor. Nivolumab o nivolumab en combinación con ipilimumab se debe suspender de forma permanente si se produce cualquier reacción adversa mediada inmunológicamente grave recurrente y ante cualquier reacción adversa mediada inmunológicamente que pueda poner en riesgo la vida. Uso de Nivolumab en pacientes con melanoma con una rápida progresión de la enfermedad: El médico debe considerar que el efecto de nivolumab puede aparecer con cierto retraso antes de iniciar el tratamiento, en pacientes cuya enfermedad progresa rápidamente. Uso de Nivolumab en pacientes con cpcnp no escamoso: El médico debe considerar que el efecto de nivolumab puede aparecer con cierto retraso antes de iniciar el tratamiento, en pacientes con factores pronósticos más pobres y/o enfermedad agresiva. En CPCNP no escamoso, se observó un mayor número de muertes dentro de los 3 meses, en nivolumab comparado con docetaxel. Los factores asociados con las muertes tempranas fueron factores pronósticos más pobres y/o una enfermedad más agresiva combinados con la baja o nula expresión tumoral de PD-L1. Neumonitis mediada inmunológicamente: Se ha observado neumonitis grave o enfermedad pulmonar intersticial, incluyendo casos mortales, con nivolumab en monoterapia o con nivolumab en combinación con ipilimumab. Se debe monitorear a los pacientes en busca de signos y síntomas de neumonitis, tales como cambios radiográficos (por ejemplo, imagen en vidrio despulido, en la base, infiltrados en parches), disnea e hipoxia. Se deben descartar etiologías infecciosas o relacionadas con la enfermedad. En caso de neumonitis Grado 3 o 4, se debe suspender permanentemente nivolumab o nivolumab en combinación con ipilimumab, y se debe iniciar tratamiento con corticosteroides a una dosis equivalente de 2 a 4 mg/kg/día de metilprednisolona. En caso de neumonitis Grado 2 (sintomática), se debe interrumpir nivolumab o nivolumab en combinación con ipilimumab, e iniciar tratamiento con corticosteroides a una dosis equivalente de 1 mg/kg/día de metilprednisolona. Una vez que se produzca la mejoría, se debe reanudar nivolumab o nivolumab en combinación con ipilimumab tras la disminución gradual de la dosis de corticosteroides. Si se produce un empeoramiento o no se observa una mejoría a pesar del inicio del tratamiento con corticosteroides, se debe aumentar la dosis equivalente de corticosteroides hasta 2 a 4 mg/kg/día de metilprednisolona y suspender permanentemente nivolumab o nivolumab en combinación con ipilimumab. Colitis mediada inmunológicamente: Se ha observado diarrea o colitis grave asociada a nivolumab en monoterapia o con nivolumab en combinación con ipilimumab. Se debe monitorear a los pacientes en relación a su diarrea y a otros síntomas relacionados con la colitis, como dolor abdominal y presencia de moco o sangre en las heces; se deben descartar etiologías infecciosas o relacionadas con la enfermedad. En caso de diarrea o colitis Grado 4, se debe suspender permanentemente nivolumab o nivolumab en combinación con ipilimumab, e iniciar tratamiento con corticosteroides a una dosis equivalente de 1 a 2 mg/kg/día de metilprednisolona. Cuando se administra nivolumab en monoterapia y se presenta diarrea o colitis Grado 3, éste se debe interrumpir e iniciar tratamiento con corticosteroides a una dosis equivalente de 1 a 2 mg/kg/día de metilprednisolona. Una vez que se observe una mejoría, reiniciar nivolumab en monoterapia tras la disminución gradual de la dosis de corticosteroides. Si se produce un empeoramiento o no se observa una mejoría a pesar del inicio del tratamiento con corticosteroides, se debe suspender permanentemente nivolumab en monoterapia. Se ha observado diarrea o colitis Grado 3 con nivolumab en combinación con ipilimumab, lo que requiere una suspensión permanente del tratamiento y el inicio de corticosteroides a una dosis equivalente de 1 a 2 mg/kg/día de metilprednisolona. En caso de diarrea o colitis Grado 2, interrumpir nivolumab o nivolumab en combinación con ipilimumab. Si persiste la diarrea o la colitis, se debe manejar con corticosteroides a una dosis equivalente de 0.5 a 1 mg/kg/día de metilprednisolona. Si se produce mejoría, reanudar el tratamiento con nivolumab o nivolumab en combinación con ipilimumab tras la disminución gradual de la dosis de corticosteroides si fuera necesario. Si se produce un empeoramiento o no se observa una mejoría a pesar del inicio del tratamiento con corticosteroides, aumentar la dosis equivalente de 1 a 2 mg/kg/día de metilprednisolona y suspender permanentemente nivolumab o nivolumab en combinación con ipilimumab. Hepatitis mediada inmunológicamente: Se ha observado hepatitis grave con nivolumab en monoterapia o con nivolumab en combinación con ipilimumab. Se debe monitorear a los pacientes en busca de signos y síntomas de hepatitis, tales como elevaciones de transaminasas y bilirrubina total. Se deben descartar etiologías infecciosas o relacionadas con la enfermedad. En caso de elevaciones de transaminasas o bilirrubina total Grado 3 o 4, se debe suspender permanentemente nivolumab o nivolumab en combinación con ipilimumab, e iniciar el tratamiento con corticosteroides a una dosis equivalente de 1 a 2 mg/kg/día de metilprednisolona. En caso de elevaciones de transaminasas o bilirrubina total Grado 2, se debe interrumpir nivolumab o nivolumab en combinación con ipilimumab. Si persisten las elevaciones en estos parámetros de laboratorio, se deben manejar con corticosteroides a una dosis de 0.5 a 1 mg/kg/día de metilprednisolona o equivalente. Una vez que se produzca la mejoría, se debe reiniciar nivolumab o nivolumab en combinación con ipilimumab tras la disminución gradual de la dosis de corticosteroides si fuera necesario. Si se produce un empeoramiento o no se observa una mejoría a pesar del inicio del tratamiento con corticosteroides, se debe aumentar la dosis equivalente de 1 a 2 mg/kg/día de metilprednisolona y suspender permanentemente nivolumab o nivolumab en combinación con ipilimumab. Nefritis e insuficiencia renal mediada inmunológicamente: Se ha observado nefritis grave e insuficiencia renal con el tratamiento con nivolumab en monoterapia o en combinación con ipilimumab. Se debe monitorear a los pacientes en busca de signos y síntomas de nefritis o insuficiencia renal. La mayoría de los pacientes presentan un aumento asintomático de la creatinina sérica. Se deben descartar etiologías relacionadas con la enfermedad. En caso de aumento de la creatinina sérica Grado 4, suspender permanentemente nivolumab o nivolumab en combinación con ipilimumab, e iniciar el tratamiento con corticosteroides a una dosis equivalente de 1 a 2 mg/kg/día de metilprednisolona. En caso de aumento de la creatinina sérica Grado 2 o 3, interrumpir nivolumab o nivolumab en combinación con ipilimumab, e iniciar tratamiento con corticosteroides a una dosis equivalente de 0.5 a 1 mg/kg/día de metilprednisolona. Una vez que se produzca la mejoría, se debe reiniciar nivolumab o nivolumab en combinación con ipilimumab tras la disminución gradual de la dosis de corticosteroides. Si se produce un empeoramiento o no se observa una mejoría a pesar del inicio del tratamiento con corticosteroides, se debe aumentar la dosis equivalente de 1 a 2 mg/kg/día de metilprednisolona y suspender permanentemente nivolumab o nivolumab en combinación con ipilimumab. Endocrinopatías mediadas inmunológicamente: Se han observado endocrinopatías graves, incluyendo hipotiroidismo, hipertiroidismo, insuficiencia suprarrenal, hipofisitis, diabetes mellitus, y cetoacidosis diabética asociadas a nivolumab en monoterapia o nivolumab en combinación con ipilimumab. Los pacientes se deben monitorear para la detección de signos y síntomas de endocrinopatías e hiperglucemias y para evaluar los cambios en la función tiroidea (al comienzo del tratamiento, periódicamente durante el tratamiento y como se ha indicado con base en su evaluación clínica). Los pacientes pueden presentar fatiga, cefalea, cambios en el estado mental, dolor abdominal, hábitos intestinales inusuales e hipotensión o síntomas no específicos que pueden parecerse a otras causas como metástasis cerebrales u otra enfermedad subyacente. A menos que otra etiología alternativa se haya identificado, los signos y síntomas de endocrinopatías se deben considerar mediados inmunológicamente. Para el hipotiroidismo sintomático, se debe interrumpir nivolumab o nivolumab en combinación con ipilimumab, y se debe iniciar tratamiento de sustitución con hormona tiroidea si fuera necesario. Para hipertiroidismo sintomático, se debe interrumpir nivolumab o nivolumab en combinación con ipilimumab, y se debe iniciar tratamiento con medicación antitiroidea si fuera necesario. También podría considerarse el tratamiento con corticosteroides a una dosis equivalente entre 1 a 2 mg/kg/día de metilprednisolona, si se sospecha de inflamación aguda de la tiroides. Una vez que se produzca una mejoría, se debe reiniciar nivolumab o nivolumab en combinación con ipilimumab tras la disminución gradual de la dosis de corticosteroides si fuera necesario. El monitoreo de la función tiroidea debe continuar para asegurar que se ha utilizado el tratamiento de sustitución hormonal adecuado. Nivolumab o nivolumab en combinación con ipilimumab se debe suspender permanentemente en caso de hipertiroidismo o hipotiroidismo que puedan poner en riesgo la vida. En caso de insuficiencia suprarrenal sintomática Grado 2, se debe interrumpir nivolumab o nivolumab en combinación con ipilimumab, y se debe iniciar tratamiento fisiológico de sustitución con corticosteroides si fuera necesario. Nivolumab o nivolumab en combinación con ipilimumab se debe suspender permanentemente en caso de insuficiencia suprarrenal grave (Grado 3) o en caso de insuficiencia suprarrenal (Grado 4) que pueda poner en riesgo la vida. El monitoreo de la función suprarrenal y los niveles hormonales deben continuar para asegurar que se ha utilizado el tratamiento de sustitución con corticosteroides adecuado. En caso de hipofisitis sintomática Grado 2 o 3, se debe interrumpir nivolumab o nivolumab en combinación con ipilimumab, y se debe iniciar tratamiento de sustitución hormonal si fuera necesario. También se podría considerar el tratamiento con corticosteroides a una dosis equivalente entre 1 a 2 mg/kg/día de metilprednisolona si se sospecha de inflamación aguda de la glándula pituitaria. Una vez que se produzca una mejoría, se debe reiniciar nivolumab o nivolumab en combinación con ipilimumab tras la disminución gradual de la dosis de corticosteroides si fuera necesario. Nivolumab o nivolumab en combinación con ipilimumab se debe suspender permanentemente en caso de hipofisitis que pueda poner en riesgo la vida (Grado 4). El monitoreo de la función de la hipófisis y de los niveles hormonales deben continuar para asegurar que se ha utilizado el tratamiento de sustitución hormonal adecuado. En caso de diabetes sintomática, se debe interrumpir nivolumab o nivolumab en combinación con ipilimumab, y se debe iniciar tratamiento de sustitución con insulina si fuera necesario. El monitoreo de la glucosa en sangre debe continuar para asegurar que se ha utilizado el tratamiento de sustitución de insulina adecuado. Nivolumab o nivolumab en combinación con ipilimumab se debe suspender permanentemente en caso de diabetes que pueda poner en riesgo la vida. Reacciones a la infusión: Se han reportado reacciones graves a la infusión en los ensayos clínicos de nivolumab o nivolumab en combinación con ipilimumab. En caso de una reacción a la infusión grave o que ponga en riesgo la vida, se debe suspender nivolumab o nivolumab en combinación con ipilimumab, y se debe administrar tratamiento médico adecuado. Los pacientes con reacciones a la infusión leves o moderadas pueden recibir nivolumab o nivolumab en combinación con ipilimumab con un estrecho monitoreo y uso de premedicación de acuerdo a las guías locales de tratamiento profiláctico de las reacciones a la infusión. Uso pediátrico: No se ha establecido la seguridad y eficacia de OPDIVO® en pacientes pediátricos. Uso geriátrico: De los 1359 pacientes aleatorizados a OPDIVO® como agente único en los estudios N Engl J Med 2015;373:123-35, N Engl J Med 2015;373:1627-39, N Engl J Med 2015;372:320-30, N Engl J Med 2015;373:1803-13 y N Engl J Med 2015;373:23-34, 39% de los pacientes tuvieron 65 años de edad o más y 9% tuvieron 75 años o más. En general, no se reportaron diferencias en la seguridad o eficacia entre los pacientes de edad avanzada y los pacientes más jóvenes. En los estudios Lancet Oncol 2015;16:375-84, Lancet Oncol 2016;17(9):1283-94, J Clin Oncol 2016;34(23):2698-2704 y N Engl J Med 2016;375(19):1856-67, no incluyeron un número suficiente de pacientes de 65 años o más para determinar si responden de manera diferente a los pacientes más jóvenes. De los 314 pacientes aleatorizados a OPDIVO® administrado con ipilimumab en el Estudio Lancet Oncol 2016;17(9):1283-94, 41% de los pacientes tuvieron 65 años o más y 11% tuvieron 75 años o más. En general, no se reportaron diferencias en la seguridad y eficacia entre los pacientes de edad avanzada y los pacientes más jóvenes. Insuficiencia renal: Basado en un análisis de farmacocinética poblacional, no está recomendado un ajuste de dosis en pacientes con insuficiencia renal. [Ver Farmacocinética y farmacodinamia, farmacocinética] Insuficiencia hepática: Basado en un análisis de farmacocinética poblacional, no está recomendado un ajuste de dosis en pacientes con insuficiencia hepática leve. OPDIVO® no ha sido estudiado en pacientes con insuficiencia hepática moderada o grave. [Ver Farmacocinética y farmacodinamia, farmacocinética]
Restricciones de uso durante el embarazo y la lactancia: Embarazo: Resumen del riesgo: Con base en su mecanismo de acción y en datos de estudios en animales, OPDIVO® puede causar daño fetal cuando se administra a mujeres embarazadas. [Ver Farmacocinética y farmacodinamia, mecanismo de acción] En estudios de reproducción animal, la administración de nivolumab a monos cynomolgus desde el inicio de la organogénesis hasta el parto aumentó la tasa de abortos y la tasa de muerte neonatal prematura. [Ver Restricciones de uso durante el embarazo y la lactancia, datos] Se sabe que la IgG4 humana cruza la barrera placentaria y nivolumab es una inmunoglobulina G4 (IgG4); por lo tanto, nivolumab puede transmitirse de la madre al feto en desarrollo. Es probable que los efectos de OPDIVO® sean mayores durante el segundo y tercer trimestres del embarazo. No hay datos en humanos que informen sobre el riesgo relacionado con el medicamento. Debe advertirse a las mujeres embarazadas del potencial riesgo para el feto. Se desconoce el antecedente del riesgo de defectos congénitos mayores y aborto para la población en la que está indicado; sin embargo, el antecedente del riesgo en la población general de Estados Unidos de defectos congénitos mayores es de 2% a 4%, y el de aborto es de 15% a 20% de los embarazos reconocidos de manera clínica. Datos: Datos en animales: Una función central de la vía PD-1/PD-L1 es conservar el embarazo al mantener la tolerancia inmunitaria materna al feto. En modelos murinos de embarazo se demostró que el bloqueo de la señalización PD-L1 afecta la tolerancia al feto y aumenta la pérdida fetal. Los efectos de nivolumab en el desarrollo prenatal y posnatal se evaluaron en monos que recibieron nivolumab dos veces a la semana desde el inicio de la organogénesis hasta el parto, con niveles de exposición entre 9 y 42 veces mayor que los alcanzados con la dosis clínica de 3 mg/kg de nivolumab (con base en el AUC). La administración de nivolumab produjo un aumento no relacionado con la dosis en los abortos espontáneos y aumentó la muerte neonatal. Con base en su mecanismo de acción, la exposición fetal a nivolumab eleva el riesgo de desarrollar trastornos mediados inmunológicamente o alterar la respuesta inmune normal y hay reportes de trastornos mediados inmunológicamente en ratones con eliminación génica de PD-1. En los lactantes sobrevivientes (18 de 32, comparados con 11 de 16 expuestos al vehículo) de monos cynomolgus tratados con nivolumab no hubo malformaciones aparentes ni se observaron efectos en los parámetros neuroconductuales, inmunitarios o de patología clínica durante el periodo posnatal de seis meses. Lactancia: Resumen del riesgo: No se sabe si OPDIVO® está presente en la leche humana. Como muchos fármacos, incluidos los anticuerpos, se excretan en la leche humana y debido a la posibilidad de reacciones adversas graves de OPDIVO® en lactantes, debe recomendarse a las mujeres que suspendan la lactancia durante el uso de OPDIVO®.
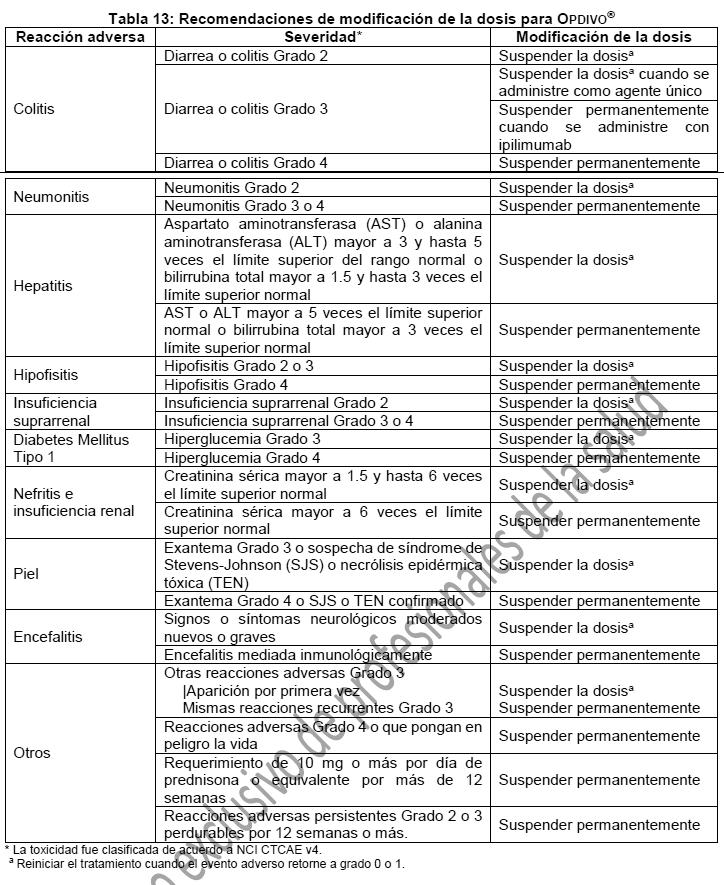
Reacciones secundarias y adversas: Neumonitis mediada inmunológicamente: OPDIVO® puede causar neumonitis mediada inmunológicamente, definida por la necesidad de usar corticosteroides y con una etiología subyacente no establecida. Se han reportado casos letales. Monitorear a los pacientes en busca de signos radiográficos y síntomas de neumonitis. Administrar corticosteroides en dosis de 1 a 2 mg/kg/día de prednisona o equivalente para la neumonitis moderada (Grado 2) o más grave (Grado 3-4), seguidos de una reducción gradual del corticosteroide. Suspender de manera permanente OPDIVO® en caso de neumonitis grave (Grado 3) o que ponga en peligro la vida (Grado 4) e interrumpir OPDIVO® hasta la resolución de la neumonitis a moderada (Grado 2). [Ver Dosis y vía de administración, modificaciones de la dosis recomendada]. OPDIVO® como agente único: En pacientes recibiendo OPDIVO® como agente único, se presentó neumonitis mediada inmunológicamente en 3.1% (61/1994) de los pacientes. La mediana de tiempo para el inicio de la neumonitis mediada inmunológicamente fue 3.5 meses (rango: 1 día a 22.3 meses). La neumonitis mediada inmunológicamente condujo a la suspensión permanente de OPDIVO® en 1.1% de los pacientes, y a la interrupción de OPDIVO® en 1.3% de los pacientes. Aproximadamente 89% de los pacientes con neumonitis recibieron dosis altas de corticosteroides (al menos 40 mg de prednisona o su equivalente por día) con una mediana de duración de 26 días (rango: 1 día a 6 meses). Se presentó una resolución completa seguida de una reducción gradual del corticosteroide en 67% de los pacientes. Aproximadamente 8% de los pacientes tuvieron una recurrencia de la neumonitis después del reinicio de OPDIVO®. OPDIVO® con ipilimumab: En pacientes recibiendo OPDIVO® con ipilimumab, se presentó neumonitis mediada inmunológicamente en 6% (25/407) de los pacientes. La mediana de tiempo para el inicio de la neumonitis mediada inmunológicamente fue 1.6 meses (rango: 24 días a 10.1 meses). La neumonitis mediada inmunológicamente condujo a la suspensión permanente o a la interrupción de OPDIVO® con ipilimumab en 2.2% y 3.7% de los pacientes respectivamente. Aproximadamente 84% de los pacientes con neumonitis recibieron dosis altas de corticosteroides (al menos 40 mg de prednisona o su equivalente por día) con una mediana de duración de 30 días (rango: 5 días a 11.8 meses). Se presentó una resolución completa en 68% de los pacientes. Aproximadamente 13% de los pacientes tuvieron una recurrencia de la neumonitis después del reinicio de OPDIVO® con ipilimumab. Colitis mediada inmunológicamente: OPDIVO® puede causar colitis mediada inmunológicamente, definida por la necesidad de usar corticosteroides y con una etiología subyacente no establecida. Monitorear a los pacientes en busca de signos y síntomas de colitis. Administrar corticosteroides en dosis de 1 a 2 mg/kg/día de prednisona o equivalente para colitis grave (Grado 3) o que ponga en riesgo la vida (Grado 4) seguido de una disminución gradual del corticosteroide. Administrar corticosteroides en dosis de 0.5 a 1 mg/kg/día de prednisona o equivalente para colitis moderada (Grado 2) con más de 5 días de duración; en caso de empeoramiento o ausencia de mejoría a pesar del inicio de los corticosteroides, aumentar la dosis a 1 a 2 mg/kg/día de prednisona o equivalente. Interrumpir OPDIVO® en caso de colitis moderada o grave (Grado 2 o 3). Suspender permanentemente OPDIVO® en caso de colitis que ponga en riesgo la vida (Grado 4) o en caso de colitis recurrente tras el reinicio de OPDIVO®. [Ver Dosis y vía de administración, modificaciones de la dosis recomendada]. Cuando se administra en combinación con ipilimumab, interrumpir OPDIVO® e ipilimumab en caso de colitis moderada (Grado 2). Suspender permanentemente OPDIVO® e ipilimumab en caso de colitis grave o que ponga en peligro la vida (Grado 3 o 4) o colitis recurrente. [Ver Dosis y vía de administración, modificaciones de la dosis recomendada]. OPDIVO® como agente único: En pacientes recibiendo OPDIVO® como agente único, se presentó colitis mediada inmunológicamente en 2.9% (58/1994) de los pacientes; la mediana del tiempo para el inicio fue 5.3 meses (rango: 2 días a 20.9 meses). La colitis mediada inmunológicamente condujo a la suspensión permanente de OPDIVO® en 0.7% de los pacientes y a la interrupción de OPDIVO® en 1% de los pacientes. Aproximadamente 91% de los pacientes con colitis recibieron dosis altas de corticosteroides (al menos 40 mg de prednisona o su equivalente por día) con una mediana de duración de 23 días (rango: 1 día a 9.3 meses). Cuatro pacientes requirieron infliximab adicionalmente a las dosis altas de corticosteroides. Se presentó una resolución completa en 74% de los pacientes. Aproximadamente 16% de los pacientes tuvieron una recurrencia de la colitis después del reinicio de OPDIVO®. OPDIVO® con ipilimumab: En pacientes recibiendo OPDIVO® con ipilimumab, se presentó colitis mediada inmunológicamente en 26% (107/407) de los pacientes incluyendo tres casos mortales. La mediana del tiempo para el inicio de la colitis mediada inmunológicamente fue 1.6 meses (rango: 3 días a 15.2 meses). La colitis mediada inmunológicamente condujo a la suspensión permanente o la interrupción de OPDIVO® con ipilimumab en 16% y 7% de los pacientes respectivamente. Aproximadamente 96% de los pacientes con colitis recibieron dosis altas de corticosteroides (al menos 40 mg de prednisona o su equivalente por día) con una mediana de duración de 1.1 meses (rango: 1 día a 12 meses). Aproximadamente 23% de los pacientes requirieron infliximab adicionalmente a las dosis altas de corticosteroides. Se presentó una resolución completa en 75% de los pacientes. Aproximadamente 28% de los pacientes tuvieron una recurrencia de la colitis después del reinicio de OPDIVO® con ipilimumab. Hepatitis mediada inmunológicamente: OPDIVO® puede causar hepatitis mediada inmunológicamente, definida por la necesidad de usar corticosteroides y con una etiología subyacente no establecida. Monitorear a los pacientes para detectar anormalidades en las pruebas hepáticas antes del tratamiento y luego de manera periódica durante éste. Administrar corticosteroides en dosis de 1 a 2 mg/kg/día de prednisona o equivalente seguido de una disminución gradual del corticosteroide en caso de elevaciones de transaminasas graves (Grado 3) o que ponen en peligro la vida (Grado 4), con o sin aumento concomitante de la bilirrubina total. Administrar corticosteroides en dosis de 0.5 a 1 mg/kg/día de prednisona o equivalente en caso de elevaciones de transaminasas moderadas (Grado 2). Interrumpir OPDIVO® en caso de hepatitis moderada mediada inmunológicamente (Grado 2) y suspender OPDIVO® de manera permanente en caso de hepatitis grave mediada inmunológicamente (Grado 3) o que pone en peligro la vida (Grado 4). [Ver Dosis y vía de administración, modificaciones de la dosis recomendada] OPDIVO® como agente único: En pacientes recibiendo OPDIVO® como agente único, se presentó hepatitis mediada inmunológicamente en 1.8% (35/1994) de los pacientes; la mediana del tiempo para el inicio fue 3.3 meses (rango: 6 días a 9 meses). La hepatitis mediada inmunológicamente condujo a la suspensión permanente de OPDIVO® en 0.7% de los pacientes y a la interrupción de OPDIVO® en 1% de los pacientes. Todos los pacientes con hepatitis recibieron dosis altas de corticosteroides (al menos 40 mg de prednisona o su equivalente) con una mediana de duración de 23 días (rango: 1 día a 2 meses). Dos pacientes requirieron ácido micofenólico adicionalmente a las dosis altas de corticosteroides. Se presentó una resolución completa en 74% de los pacientes. Aproximadamente 29% de los pacientes tuvieron una recurrencia de la hepatitis después del reinicio de OPDIVO®. OPDIVO® con ipilimumab: En pacientes recibiendo OPDIVO® con ipilimumab, se presentó hepatitis mediada inmunológicamente en 13% (51/407) de los pacientes; la mediana del tiempo para el inicio fue 2.1 meses (rango: 15 días a 11 meses). La hepatitis mediada inmunológicamente condujo a una suspensión permanente o interrupción de OPDIVO® con ipilimumab en 6% y 5% de los pacientes respectivamente. Aproximadamente 92% de los pacientes con hepatitis recibieron dosis altas de corticosteroides (al menos 40 mg de prednisona o su equivalente por día) con una mediana de duración de 1.1 meses (rango: 1 día a 13.2 meses). Se presentó una resolución completa en 75% de los pacientes. Aproximadamente 11% de los pacientes tuvieron una recurrencia de la hepatitis después del reinicio de OPDIVO® con ipilimumab. Endocrinopatías mediadas inmunológicamente: Hipofisitis: OPDIVO® puede causar hipofisitis mediada inmunológicamente. Monitorear a los pacientes en busca de signos y síntomas de hipofisitis. Administrar terapia de reemplazo hormonal de acuerdo a indicación clínica y corticosteroides en dosis de 1 mg/kg/día de prednisona o equivalente seguido de una disminución gradual del corticosteroide para la hipofisitis moderada (Grado 2) o más grave. Interrumpir OPDIVO® en caso de hipofisitis moderada (Grado 2) o grave (Grado 3). Suspender OPDIVO® de manera permanente si hay hipofisitis que ponga en peligro la vida (Grado 4). [Ver Dosis y vía de administración, modificaciones de la dosis recomendada]. En pacientes recibiendo OPDIVO® como agente único, se presentó hipofisitis en 0.6% (12/1994) de los pacientes; la mediana de tiempo para el inicio fue 4.9 meses (rango: 1.4 a 11 meses). La hipofisitis condujo a la suspensión permanente de OPDIVO® en 0.1% de los pacientes y a la interrupción de OPDIVO® en 0.2% de los pacientes. Aproximadamente 67% de los pacientes con hipofisitis recibieron terapia de reemplazo hormonal y 33% recibieron dosis altas de corticosteroides (al menos 40 mg de prednisona o equivalente por día) por una mediana de duración de 14 días (rango: 5 a 26 días). En pacientes recibiendo OPDIVO® con ipilimumab, se presentó hipofisitis en 9% (36/407) de los pacientes; la mediana de tiempo para el inicio fue 2.7 meses (rango: 27 días a 5.5 meses). La hipofisitis condujo a una suspensión permanente o interrupción de OPDIVO® con ipilimumab 1.0% y 3.9% de los pacientes respectivamente. Aproximadamente 75% de los pacientes con hipofisitis recibieron terapia de reemplazo hormonal y 56% recibieron dosis altas de corticosteroides (al menos 40 mg de prednisona o equivalente por día) por una mediana de duración de 19 días (rango: 1 día a 2.0 meses). Insuficiencia suprarrenal: OPDIVO® puede causar insuficiencia suprarrenal mediada inmunológicamente. Monitorear a los pacientes en busca de signos y síntomas de insuficiencia suprarrenal. Administrar corticosteroides en dosis de 1 a 2 mg/kg/día de prednisona o equivalente para insuficiencia suprarrenal grave (Grado 3) o que ponga en peligro la vida (Grado 4), seguido de una disminución gradual del corticosteroide. Interrumpir OPDIVO® si la insuficiencia suprarrenal es moderada (Grado 2) y suspenderlo de manera permanente si es grave (Grado 3) o pone en peligro la vida (Grado 4). [Ver Dosis y vía de administración, modificaciones de la dosis recomendada]. En pacientes recibiendo OPDIVO® como agente único se presentó insuficiencia suprarrenal en 1% (20/1994) de los pacientes y la mediana de tiempo para el inicio fue 4.3 meses (rango: 15 días a 21 meses). La insuficiencia suprarrenal condujo a la suspensión permanente de OPDIVO® en 0.1% de los pacientes y a la interrupción de OPDIVO® en 0.5% de los pacientes. Aproximadamente 85% de los pacientes con insuficiencia suprarrenal recibieron terapia de reemplazo hormonal y 25% recibieron dosis altas de corticosteroides (al menos 40 mg de prednisona o equivalente por día) por una mediana de duración de 11 días (rango: 1 día a 1 mes). En pacientes recibiendo OPDIVO® con ipilimumab, se presentó insuficiencia suprarrenal en 5% (21/407) de los pacientes, la mediana de tiempo para el inicio fue 3 meses (rango: 21 días a 9.4 meses). La insuficiencia suprarrenal condujo a la suspensión permanente o interrupción de OPDIVO® con ipilimumab en 0.5% y 1.7% de los pacientes respectivamente. Aproximadamente 57% de los pacientes con insuficiencia suprarrenal recibieron terapia de reemplazo hormonal y 33% recibieron dosis altas de corticosteroides (al menos 40 mg de prednisona o equivalente por día) por una mediana de duración de 9 días (rango: 1 día a 2.7 meses). Hipotiroidismo e Hipertiroidismo: OPDIVO® puede causar trastornos tiroideos autoinmunes. Monitorear la función tiroidea antes de iniciar el tratamiento y luego periódicamente durante el tratamiento con OPDIVO®. Administrar terapia de reemplazo hormonal en caso de hipotiroidismo. Iniciar el tratamiento médico para controlar el hipertiroidismo. No se recomiendan ajustes en la dosis de OPDIVO® en presencia de hipotiroidismo o hipertiroidismo. En pacientes recibiendo OPDIVO® como agente único, se presentó hipotiroidismo o tiroiditis resultando en hipotiroidismo en 9% (171/1994) de los pacientes; la mediana de tiempo para el inicio fue 2.9 meses (rango: 1 día a 16.6 meses). Aproximadamente 79% de los pacientes con hipotiroidismo recibieron levotiroxina y 4% también requirieron corticosteroides. Se presentó una resolución en 35% de los