PAMIGEN®
ADIUM
Denominación genérica: Gemcitabina.
Forma farmacéutica y formulación: Solución. Cada frasco ámpula con liofilizado contiene: Clorhidrato de Gemcitabina equivalente a 200 mg y 1000 mg de Gemcitabina. Excipiente c.b.p 1 frasco ámpula.
Indicaciones terapéuticas: PAMIGEN® (Gemcitabina), está indicado como terapia de primera línea para el tratamiento de pacientes con cáncer pulmonar de células no pequeñas localmente avanzado o metastásico solo o en combinación con cisplatino. PAMIGEN® está indicado para el tratamiento de primera línea en pacientes con adenocarcinoma de páncreas localmente avanzado o metastásico. PAMIGEN® está indicado para pacientes con cáncer pancreático refractario a 5-FU. PAMIGEN® en combinación con cisplatino está indicado en el tratamiento de pacientes con cáncer de vejiga avanzado y/o metastásico y en pacientes con carcinoma de células superficiales de vejiga, en refractariedad a la terapia con BCG. PAMIGEN®, indicado en combinación con paclitaxel para el tratamiento de pacientes con cáncer de mama no resecable, localmente recurrente o metastásico, que hayan recaído luego de recibir quimioterapia adjuvante o neoadjuvante basada en antraciclinas. PAMIGEN® está indicado solo o en en combinación para el tratamiento de pacientes con carcinoma epitelial de ovario recurrente que han recaído como mínimo 6 meses después del tratamiento con platino. PAMIGEN® está indicado en el tratamiento de primera línea del cáncer cervicouterino localmente avanzado o metastásico. PAMIGEN®, indicado en el tratamiento de los pacientes con cáncer del tracto biliar.
Farmacocinética y farmacodinamia: Farmacocinética: Gemcitabina es rápidamente eliminada del plasma, principalmente por metabolismo de un metabolito inactivo, la 2'-desoxi-2',2'-difluorouridina (dFdU). Menos del 10% de una dosis intravenosa se recupera en la orina como Gemcitabina intacta. La Gemcitabina y dFdU son los únicos compuestos hallados en el plasma y constituyen el 99% del material recuperado en orina. La unión de la Gemcitabina a proteínas plasmáticas es muy baja < 10%. Los análisis de los estudios farmacocinéticos poblacionales de dosis simple y múltiple han demostrado que el volumen de distribución de Gemcitabina está significativamente influido por el sexo. El aclaramiento sistémico se encuentra afectado por el sexo y la edad. Estos efectos determinan diferencias en las concentraciones plasmáticas de Gemcitabina y su velocidad de eliminación (vida media) de la circulación sistémica. El aclaramiento sistémico se encuentra dentro de un rango aproximado de 30 L/hr/m2 a 90 L/hr/m2. A la velocidad de infusión recomendada, su vida media oscila entre 32 a 94 minutos, dependiendo de la edad y el sexo. Farmacodinamia: La Gemcitabina es específica en el ciclo celular actuando en la fase S, eliminando las células que se encuentran en síntesis de ADN (fase S) y bajo ciertas condiciones, bloqueando la progresión de las células a través de la fase de unión G1/S. La Gemcitabina (dFdC) se metaboliza intracelularmente por nucleósidoquinasas a difosfato (dFdCDP) y trifosfato (dFdCTP) nucleósidos activos. La acción citotóxica de la Gemcitabina parece ser debida a la inhibición de la síntesis del ADN por dos acciones del dFdCDP y del dFdCTP. Primero, el dFdCDP inhibe a la ribonucleótido-reductasa que es la única responsable de catalizar las reacciones que generan los trifosfatos desoxinucleósidos para la síntesis del ADN. La inhibición de esta enzima por el dFdCDP, causa una reducción en las concentraciones de desoxinucleósidos en general, y especialmente en aquella del dCTP. Segundo, el dFdCTP compite con el dCTP por la incorporación al ADN (auto-potenciación). De este modo, la reducción en la concentración intracelular de dCTP potencia la incorporación de dFdCTP al ADN. El ADN polimerasa epsilon es fundamentalmente incapaz de remover la Gemcitabina y reparar los crecientes filamentos del ADN. Luego que la Gemcitabina es incorporada al ADN, un nucleótido adicional se agrega a los filamentos crecientes de ADN. Después de este agregado, se produce esencialmente una inhibición completa en la síntesis posterior del ADN (terminación encubierta de la cadena). Después de la incorporación al ADN, la Gemcitabina aparece luego para inducir el proceso de muerte celular programada conocido como apoptosis.
Contraindicaciones: PAMIGEN® está contraindicado en pacientes con hipersensibilidad conocida al medicamento.
Precauciones generales: Debido a que PAMIGEN® puede inducir supresión en la médula ósea, los pacientes que reciban terapia con este fármaco deben ser monitoreados continuamente en el conteo sanguíneo de glóbulos rojos y glóbulos blancos. La prolongación en el tiempo de infusión y aumento en la intensidad de dosis puede incrementar los efectos de toxicidad. Los pacientes comprometidos por toxicidad debida al fármaco pueden requerir tratamiento.
Precauciones y restricciones de uso durante el embarazo y la lactancia: El uso de Gemcitabina durante el embarazo debe evitarse por el potencial peligro para el feto. La evaluación de estudios con animales de experimentación ha demostrado toxicidad en la reproducción, por ej. defectos de nacimiento u otros efectos en el desarrollo del embrión o feto, el curso de gestación o desarrollo peri y postnatal. El uso de Gemcitabina debe evitarse en mujeres en período de lactancia por el potencial peligro para el lactante.
Reacciones secundarias y adversas: Hematológicas: Como la Gemcitabina es un supresor medular óseo, puede inducir a anemia, leucopenia y trombocitopenia, como resultado de su administración. También se informa comúnmente neutropenia febril. Gastrointestinales: Las anormalidades en las pruebas de función hepática son muy comunes, pero éstas son habitualmente leves, no progresivas y ocasionalmente se necesita suspender el tratamiento. No obstante, la Gemcitabina deberá ser empleada con precaución en pacientes con función hepática deteriorada. Náusea acompañada de vómito se presentan en forma frecuente. Este efecto adverso raramente resulta limitante de la dosis, y es fácilmente manejable con antieméticos habituales; diarrea y estomatitis también han sido informadas con frecuencia. Renales: Se ha informado en forma frecuente proteinuria moderada y hematuria. Piel y tegumentos: Se ha observado rash, frecuentemente asociado con prurito. El rash es por lo general moderado. También se ha reportado alopecia con frecuencia (usualmente mínima caída del cabello). Respiratorias: Disnea ha sido reportada con frecuencia. En raras ocasiones se ha reportado broncoespasmo, posterior a la infusión de Gemcitabina. Ocasionalmente se ha reportado neumonitis intersticial. En raras ocasiones se han reportado efectos pulmonares, a veces severos (como edema pulmonar o síndrome de distrés respiratorio del adulto) en asociación a la terapia con Gemcitabina. Si se desarrollaran estos efectos, se debe considerar la discontinuación del tratamiento con Gemcitabina. El uso temprano de medidas de apoyo puede mejorar la condición. Genito-urinarias: Se ha informado raramente de hallazgos clínicos consistentes con Síndrome Urémico Hemolítico en pacientes que reciben Gemcitabina. La Gemcitabina debe discontinuarse a los primeros síntomas de cualquier evidencia microangiopática de anemia hemolítica, tales como la rápida caída de la hemoglobina concomitante a trombocitopenia, elevación de la bilirrubina sérica, creatinina sérica, nitrógeno ureico o LDH. La insuficiencia renal puede no ser reversible aún con la discontinuación de la terapia y puede ser necesaria la diálisis. Generales: Se ha informado sobre sintomatología similar a la gripe. Los síntomas más comúnmente informados son: fiebre, dolor de cabeza, dolor de espalda, escalofríos, mialgia, astenia y anorexia; de igual manera: tos, rinitis, malestar, sudoración e insomnio. Se han reportado en muy raras ocasiones reacciones anafilácticas. Se ha reportado toxicidad por radiación (ver sección de interacciones). Cardiovasculares: Se ha informado frecuentemente sobre edema/edema periférico. Unos pocos casos de hipotensión han sido informados. Se han reportado casos de infarto de miocardio, insuficiencia cardiaca congestiva y arritmia, pero no existe una evidencia clara de que la Gemcitabina cause toxicidad cardiaca. Vasculares: Muy rara vez se han informado signos clínicos de vasculitis periférica y gangrena. Piel y tegumentos: Muy rara vez se han informado reacciones cutáneas severas, incluidas descamación y erupciones bulosas. Hepatobiliares: Raramente se han informado pruebas de la función hepática con anormalidades que incluyan aumentos en los niveles de aspartato aminotransferasa (AST), alanina aminotransferasa (ALT), gammaglutamil transferasa (GGT), fosfatasa alcalina y bilirrubina. Se han informado reacciones de hipersensibilización a la radiación.
Interacciones medicamentosas y de otro género: Radioterapia concomitante (administrada conjuntamente o con menos de 7 días de diferencia): La toxicidad asociada con esta terapia multimodalidad depende de muchos factores diferentes, que incluyen la dosis de Gemcitabina, la frecuencia de la administración de Gemcitabina, la dosis de radiación, la técnica para la planificación de radioterapia, el tejido blanco y el volumen objetivo. Los estudios preclínicos y clínicos demostraron que la Gemcitabina tiene actividad radiosensibilizante. En un solo estudio, donde se administró dosis de 1000 mg/m2 de Gemcitabina durante 6 semanas consecutivas como máximo, en forma concomitante con radiación terapéutica torácica a pacientes con cáncer de pulmón de células no pequeñas, se observó toxicidad significativa con posible riesgo de vida en la forma de mucositis severa, en especial esofagitis, y neumonitis, en particular en pacientes que recibían grandes volúmenes de radioterapia [mediana del volumen de tratamiento 4.7 95 cm3]. Estudios efectuados con posterioridad sugirieron que es factible administración concomitante de Gemcitabina en dosis más bajas y radioterapia con toxicidad previsible, por ejemplo, en un estudio de fase II en cáncer de pulmón de células no pequeñas, se administraron dosis de radiación torácica de 66Gy con Gemcitabina (600 mg/m2, cuatro veces) y cisplatino (80 mg/m2, dos veces) durante 6 semanas. Diversos estudios de fase I y II demostraron que es posible administrar 300 mg/m2/semana de Gemcitabina como agente único junto con radioterapia en pacientes con cáncer de pulmón de células no pequeñas y con cáncer de páncreas. Aún no se ha determinado cuál es el régimen óptimo para la administración segura de Gemcitabina con dosis terapéuticas de radiación en todo tipo de tumor. Radioterapia secuencial (administrada con más de 7 días de diferencia): El análisis de datos no indica ningún aumento de la toxicidad cuando la Gemcitabina es administrada con una diferencia mayor de 7 días antes o 7 después de la radiación, salvo hipersensibilidad a la radiación. Los datos sugieren que se puede reiniciar la administración de Gemcitabina después de que se hayan resuelto los efectos agudos de la radiación o, por lo menos, una semana después de la radiación. Se han informado lesiones por radiación en tejido blando (por ej. esofagitis, colitis y neumonitis) asociada con el uso tanto simultáneo como no simultáneo de Gemcitabina.
Alteraciones en los resultados de pruebas de laboratorio: Deberá monitorearse el recuento de plaquetas, leucocitos y granulocitos en pacientes que reciben Gemcitabina antes de cada dosis. Debe tenerse en cuenta la suspensión o modificación de la terapia cuando se detecte depresión medular inducida por la droga. Se deben realizar pruebas de laboratorio de función renal y hepática en forma periódica.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: La Gemcitabina produjo daño citogenético en un ensayo in vivo. La Gemcitabina induce mutación precoz en ensayos in vitro en linfoma de ratón (L5178Y). La Gemcitabina ha causado una hipoespermatogénesis dosis reversible y esquema de dosificación dependiente en ratones machos. Aunque los estudios en animales han demostrado un efecto de la Gemcitabina sobre la fertilidad masculina, no se ha observado ningún efecto en la fertilidad femenina. No se han realizado estudios a largo plazo en animales para evaluar el potencial carcinogénico de la Gemcitabina.
Dosis y vía de administración: Cáncer de Páncreas: Como agente único: PAMIGEN® debe ser administrado por infusión intravenosa, una dosis de 1000 mg/m2 durante 30 minutos, una vez a la semana durante 7 semanas, seguida de una semana de descanso. Las dosis siguientes deberán ser de 1 vez por semana durante 3 semanas y una de descanso. Antes de cada dosis deben monitorearse el recuento de plaquetas, leucocitos y granulocitosis y, si fuera necesario, la dosis de PAMIGEN® debe ser suspendida o reducida de acuerdo a la toxicidad hematológica observada.
Deberán monitorearse las funciones hepática y renal para detectar toxicidad no hematológica. La dosificación debe ser suspendida hasta que la toxicidad sea resuelta según la opinión del médico tratante. Cáncer del Tracto Biliar: En combinación: La Gemcitabina en combinación con cisplatino se recomienda utilizando cisplatino 70 mg/m<<2 administrado en el día 1 como una infusión intravenosa, seguido por Gemcitabina 1250 mg/m2 administrada en los días 1 y 8, de cada ciclo de 21 días, en una infusión intravenosa de 30 minutos. Este ciclo de 3 semanas es entonces repetido. Como agente único: La dosis recomendada de Gemcitabina es de 1000 mg/m2, administrada en una infusión intravenosa de 30 minutos. La dosis deber ser repetida cada semana por tres semanas seguidas de un periodo de descanso de una semana. Este ciclo de cuatro semanas debe entonces repetirse. Carcinoma de Mama: Infusión intravenosa (no más de 30 minutos), 1250 mg/m2 los días 1 y 8 de cada ciclo de 21 días. Paclitaxel debe ser administrado a 175 mg/m2 el día 1 como una infusión intravenosa de 3 horas antes de la administración de Gemcitabina. Modificación de la dosis por toxicidad hematológica. Los ajustes de la dosis están basados sobre recuentos de leucocitos y plaquetas tomados el día 8 del tratamiento. La dosis de Gemcitabina debe ser modificada de acuerdo con las siguientes directivas si se descubre supresión medular. Los pacientes deben tener una cuenta absoluta de granulocitos de cuando menos 1500 (x106/L) antes de iniciar la combinación de Gemcitabina + Paclitaxel.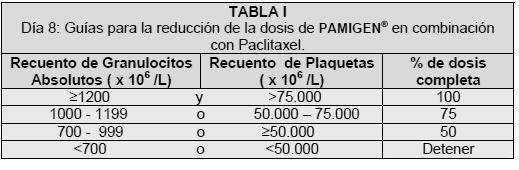
Modificación de dosis para toxicidad no hematológica severa (grado 3 ó 4). Gemcitabina debe ser detenida o reducida en un 50% dependiendo del criterio del médico tratante para toxicidad no hematológica severa (grado 3 ó 4), con excepción de alopecia y náusea/vómitos. Para el ajuste de la dosis de paclitaxel, ver la información de ese producto. La dosis recomendada de Gemcitabina como agente único es de 1000 a 1200 mg/m2 administrada en una infusión durante 30 minutos, en los días 1, 8 y 15 de cada ciclo de 28 días. Este ciclo de cuatro semanas debe entonces repetirse. La dosis en cada ciclo o dentro de un mismo ciclo se puede reducir en función de la toxicidad experimentada por el paciente. Cáncer de Pulmón de células no-pequeñas: Esquema de tres semanas en combinación con platinos: PAMIGEN® debe administrarse por vía intravenosa a 1,250 mg/m2 durante 30 minutos, en los días 1 y 8 de cada ciclo de 21 días. El cisplatino debe administrarse por vía intravenosa a una dosis de 100 mg/m2 en el día 1, después de la infusión de Gemcitabina. La dosis en cada ciclo o dentro de un mismo ciclo se reduce en función de la toxicidad experimentada por el paciente. PAMIGEN® en combinación con carboplatino se recomienda utilizando una dosis de Gemcitabina de 1000 ó 1200 mg/m2, administrada en los días 1 y 8 de cada ciclo de 21 días como una infusión intravenosa en 30 minutos. Después de Gemcitabina se administrará el carboplatino en el día 1 en forma consistente con un objetivo de área bajo la curva (AUC) de 5 mg/ml•min. Esquema de cuatro semanas en combinación con platinos: PAMIGEN® debe administrarse por vía intravenosa a 1000 mg/m2 durante 30 minutos, en los días 1, 8 y 15 de cada ciclo de 28 días. El cisplatino debe administrase por vía intravenosa a una dosis de 100 mg/m2 en el día 1, después de la infusión de Gemcitabina. La dosis en cada ciclo o dentro de un mismo ciclo se reduce en función de la toxicidad experimentada por el paciente. Esquema como agente único: La dosis recomendada de Gemcitabina es de 1000 mg/m2, administrada en una infusión intravenosa de 30 minutos. Esta debe ser repetida una vez a la semana por tres semanas, seguidas de un período de descanso de una semana. Este ciclo de cuatro semanas deberá ser repetido posteriormente. La dosis en cada ciclo o dentro de un mismo ciclo se reduce en función de la toxicidad experimentada por el paciente. Cáncer de Ovario: En combinación Gemcitabina debe ser administrado de forma intravenosa a una dosis de 1000 mg/m2 durante 30 minutos en los días 1 y 8 de cada ciclo de 21 días. El carboplatino debe ser administrado por vía intravenosa en el día 1, en forma consistente, con un objetivo de área bajo la curva (AUC) de 4 mg/ml < 195 > min luego de la administración de Gemcitabina. Los pacientes deben ser monitoreados antes de cada dosis por medio de un recuento de glóbulos rojos y blancos, incluyendo recuentos diferenciales. Los pacientes deben tener un recuento absoluto de granulocitos ≥ 1500 x 106/L y un recuento de plaquetas ≥ 100.000 x 106 /L antes de cada ciclo. Como agente único: La dosis recomendada de Gemcitabina es de 800 a 1250 mg/m2 administrada en una infusión durante 30 minutos, en los días 1, 8 y 15 de cada ciclo de 28 días. Este ciclo de cuatro semanas debe entonces repetirse. Modificaciones en las Dosis: Los ajustes de dosis de Gemcitabina para toxicidad hematológica dentro de un ciclo de tratamiento, están basados en los recuentos de granulocitos y plaquetas tomados el día 8 de la terapia. Si se detecta supresión de la médula, se debe modificar la dosis de acuerdo a las instrucciones de la Tabla II.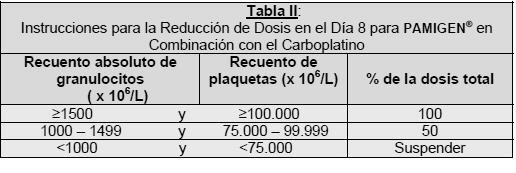
En general, para toxicidad no hematológica severa (Grado 3 ó 4), excepto nausea y vómitos, la terapia con Gemcitabina debe ser detenida o disminuida en un 50%, dependiendo del criterio del médico tratante. Para ajustes en la dosis de carboplatino, se debe ver la información para prescribir. El ajuste de dosis de Gemcitabina en combinación con el carboplatino para ciclos subsiguientes, se basa en la toxicidad observada. La dosis de Gemcitabina en ciclos subsiguientes debe reducirse a 800 mg/m2 los días 1 y 8, en caso de presentarse cualquiera de las toxicidades hematológicas: Recuentos absolutos de granulocitos < 500 x 106/L por más de 5 días. Recuentos absolutos de granulocitos < 100 x 106/L por más de 3 días. Neutropenia Febril. Plaquetas < 25.000 x 106/L. Demora del ciclo de más de una semana debido a la toxicidad. Cáncer de Vejiga: La dosis recomendada de Gemcitabina es de 1000 mg/m2 administrada en una infusión durante 30 minutos, en los días 1, 8 y 15 de cada ciclo de 28 días. El cisplatino debe administrase por vía intravenosa a una dosis de 70 mg/m2 en el día 1, después de la infusión de Gemcitabina, o en el día 2 de 12 cada ciclo de 28 días. Este ciclo de cuatro días debe entonces repetirse. La dosis en cada ciclo o dentro de un mismo ciclo se reduce en función de la toxicidad experimentada por el paciente. Un estudio clínico mostró mayor mielosupresión cuando el cisplatino se utilizó en dosis de 100 mg/m2. Uso intravesical. La dosis recomendada para el uso intravesical de Gemcitabina en cáncer superficial de vejiga es de 2000 mg, diluidos, ya sea en 100 ó 50 ml de solución salina (concentración de 20 ó 40 mg/ml). El medicamento debe permanecer intravesicalmente por espacio de 60 minutos, una vez a la semana, hasta por 6 semanas consecutivas. Las concentraciones no deben exceder 40 mg/ml y las reducciones de la dosis pueden ser aplicadas con base en la toxicidad experimentada por el paciente. Cáncer de Cérvix: Tratamiento Neoadyuvante y Enfermedad Metastásica. La Gemcitabina en combinación con cisplatino se recomienda utilizando cisplatino 70 mg/m2 administrado en el día 1, seguido por Gemcitabina 1250 mg/m2 administrado en los días 1 y 8, cada 21 días. Este ciclo de 3 semanas es entonces repetido. Terapia concomitante con radiación: La Gemcitabina en combinación con cisplatino se recomienda utilizando cisplatino 40 mg/m2 seguido inmediatamente por Gemcitabina 125 mg/m2 por infusión intravenosa una vez a la semana por 6 semanas (días 1, 8, 15, 22, 29 y 36), 1 a 2 horas antes de la radioterapia pélvica concomitante. La radioterapia debe consistir de 50.4 Gy mediante radiación externa administrados al campo completo de radiación en 28 fracciones, 1.8 Gy/día, 5 días/semana, durante las 6 semanas de quimioterapia. Control de dosificación: Insuficiencia hepática y renal: La Gemcitabina deberá ser empleada con precaución en pacientes con insuficiencia hepática o con función renal deteriorada. Una insuficiencia renal moderada (tasa de filtración glomerular de 30 a 80 mL/min) no ha tenido un efecto consistente y significativo sobre la farmacocinética de la Gemcitabina. Pacientes en edad avanzada: No hay evidencia que sugiera que los ajustes de las dosis son necesarios en pacientes de edad avanzada, aunque la depuración y vida media de la Gemcitabina resultan afectadas por la edad. Niños: La Gemcitabina no ha sido estudiada en niños.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: No existe un antídoto para la sobredosis de Gemcitabina. Las dosis tan altas como 5,7 g/m2 han sido administradas mediante infusión IV cerca de 30 minutos, cada dos semanas con toxicidad clínicamente aceptable. En el caso de sospecha de sobredosis, deberá monitorearse al paciente con recuentos de sangre adecuados y deberá recibir terapia de apoyo, en la forma que resulte necesaria.
Presentación(es): Caja con 1 frasco ámpula con liofilizado con 200 mg. Caja con 1 frasco ámpula con liofilizado con 1000 mg.
Recomendaciones sobre almacenamiento: Consérvese a temperatura ambiente a no más de 30°C. Desechar si no se utiliza en un período de 2 horas después de haberse reconstituido. Si no se administra todo el producto deséchese el sobrante.
Leyendas de protección: Vía de administración: Intravenosa. Dosis: La que el médico señale. Léase instructivo anexo. Su venta requiere receta médica. Este medicamento es de alto riesgo. No se deje al alcance de los niños. Este medicamento solo debe ser administrado bajo la supervisión de médicos especialistas en Oncología y con experiencia en quimioterapia antineoplásica. No se administre si el cierre ha sido violado. No se administre si la solución no es transparente, si contiene partículas en suspensión o sedimentos. No se use en el embarazo y lactancia. Literatura exclusiva para médicos.
Nombre y domicilio del laboratorio: Hecho en Paraguay por: FARMACEUTICA PARAGUAYA, S.A. Waldino Lovera esquina Del Carmen, Fernando de la Mora, Paraguay. Distribuido en México por: ASOFARMA DE MEXICO, S.A. DE C.V. Calz. México-Xochimilco N° 43, Col. San Lorenzo Huipulco, C.P. 14370, Delegación. Tlalpan, D.F. México.
Clave de IPPA: 113300CT050566/ Oct11