PRADAXAR®
BOEHRINGER PM
Denominación genérica: Dabigatrán.
Forma farmacéutica y formulación: Cápsulas. Cada cápsula contiene: dabigatrán etexilato mesilato equivalente a 75 o 110mg de etexilato de dabigatrán. Excipiente cbp 1 cápsula.
Indicaciones terapéuticas: Prevención de eventos tromboembólicos venosos en pacientes que han sido sometidos a cirugía ortopédica mayor.
Farmacocinética y farmacodinamia: Posterior a la administración oral de etexilato de dabigatrán en voluntarios sanos, el perfil farmacocinético de dabigatrán en plasma se caracterizó por un rápido incremento en las concentraciones plasmáticas alcanzando la Cmáx en un plazo de 0,5 a 2,0 horas posterior a su administración. Posterior a la Cmáx, se observó que las concentraciones de dabigatrán en plasma mostraron una disminución biexponencial con una vida media terminal de aproximadamente 14 a 17 horas en sujetos jóvenes sanos y de 12 a 14 horas en sujetos ancianos. La vida media fue independiente de la dosis. La Cmáx y el área bajo la curva de la relación concentración-tiempo en plasma fueron proporcionales a la dosis. Los alimentos no afectan la biodisponibilidad de etexilato de dabigatrán, pero retrasa la obtención de concentraciones plasmáticas pico por 2 horas. La biodisponibilidad absoluta de dabigatrán seguida a su administración oral fue aproximadamente del 6,5%. En un estudio que evaluó la absorción postoperatoria de etexilato de dabigatrán, 1 a 3 horas posteriores a la cirugía, demostró una absorción relativamente lenta comparada con la de los voluntarios sanos, observándose un perfil plano de concentración plasmática-tiempo sin concentraciones plasmáticas máximas altas. Las concentraciones plasmáticas máximas se alcanzan a las 6 horas de la administración, o a las 7 a 9 horas luego de la cirugía. Sin embargo, se nota que los factores contribuyentes como la anestesia, la paresia gastrointestinal y los efectos quirúrgicos significarán que una proporción de pacientes experimentará un retraso en la absorción independiente de la formulación oral del fármaco. Aunque este estudio no predice si el deterioro en la absorción persiste con las dosis subsiguientes, se demostró en un estudio posterior que la absorción lenta y retrasada está presente habitualmente sólo en el día de la cirugía. En los días subsiguientes la absorción de dabigatrán es rápida con concentraciones plasmáticas máximas alcanzadas a las 2 horas después de la administración del fármaco. El metabolismo y la excreción de dabigatrán se estudiaron seguidos de una dosis única intravenosa de dabigatrán radiomarcado en hombres sanos. Después de una dosis intravenosa, la radioactividad derivada de dabigatrán se eliminó primariamente en la orina (85%). La excreción fecal representó el 6% de la dosis administrada. La recuperación de la radioactividad total fue del 88 al 94% de la dosis administrada 168 horas posteriores a la dosis. Después de la administración oral, etexilato de dabigatrán se convierte rápida y completamente en dabigatrán, que es la forma activa en plasma. El clivaje del profármaco de etexilato de dabigatrán por medio de la hidrólisis catalizada por esterasa al principio activo dabigatrán es la reacción metabólica predominante. Dabigatrán es conjugado formando acil-glucurónidos farmacológicamente activos. Existen cuatro isómeros posicionales, 1-0, 2-0, 3-0, 4-0- acil-glucurónico, cada uno representando menos del 10% de dabigatrán total en el plasma. Trazas de otros metabolitos fueron solamente detectables con métodos analíticos de alta sensibilidad. Dabigatrán se elimina primariamente en forma intacta en la orina, a un índice de aproximadamente 100 ml/min correspondiendo al índice de filtrado glomerular. Se observó una baja unión de dabigatrán a proteínas plasmáticas humanas (34-35%), independiente de la concentración. El volumen de distribución de dabigatrán de 60 a 70 litros excedió el volumen del agua corporal total, indicando una distribución moderada de dabigatrán en los tejidos. Poblaciones especiales: insuficiencia renal: la exposición de dabigatrán después de la administración oral a etexilato de dabigatrán es aproximadamente de 2,7 veces más en voluntarios con insuficiencia renal moderada (aclaramiento de creatinina entre 30-50 ml/min) que en quienes no tienen insuficiencia renal. En un número pequeño de voluntarios con insuficiencia renal severa (aclaramiento de creatinina de 10 a 30 ml/min), la exposición (AUC) a dabigatrán fue aproximadamente 6 veces más alta, con vida media aproximadamente 2 veces más larga que la observada en la población sin insuficiencia renal. Pacientes ancianos: estudios farmacocinéticas específicos en personas ancianas mostraron un incremento de 40 a 60% en el AUC y más del 25% en la Cmáx comparado con sujetos jóvenes. En la población en la que se basaron los estudios farmacocinéticos fueron evaluados los parámetros farmacocinéticos de dabigatrán después de dosis repetidas (pacientes mayores de 88 años). Se observó que el incremento de la exposición a dabigatrán se correlaciona con la reducción en el aclaramiento de creatinina relacionada con la edad. Insuficiencia hepática: no se observaron cambios en 12 pacientes expuestos a dabigatrán con insuficiencia hepática moderada (Child-Pugh B) comparados con 12 controles. Peso corporal: se llevaron a cabo estudios farmacocinéticos que evaluaron los parámetros de dabigatrán en pacientes de 48 a 120 kg de peso corporal. El peso corporal tuvo un menor efecto en el aclaramiento de dabigatrán resultando en una mayor exposición en pacientes con menor peso corporal (BISTRO II). Género: no existieron diferencias en los estudios clínicos fase 3 para los datos de seguridad y eficacia entre hombres y mujeres. La exposición al medicamento en pacientes mujeres es del 40% al 50% más elevada que en pacientes masculinos, sin embargo no se recomienda ningún ajuste de dosis (BISTRO II). Origen étnico: la farmacocinética de dabigatrán fue investigada en voluntarios caucásicos y japoneses tanto en dosis única como en dosis múltiples. El origen étnico no afecta la farmacocinética de dabigatrán de manera clínicamente relevante. No se encuentran disponibles datos farmacocinéticos en pacientes de raza negra. Interacciones farmacocinéticas: estudios de interacción in vitro no mostraron ninguna inhibición o inducción de las principales isoenzimas o del citrocromo P450. Esto ha sido confirmado en estudios in vivo con voluntarios sanos, en quienes no se ha observado ninguna interacción entre este tratamiento y los siguientes fármacos: atorvastatina (CYP3A4), digoxina (P-gp transportador) y diclofenaco (CYP2C9). La exposición de dabigatrán en sujetos sanos fue incrementada en 60% en presencia de amiodarona. El análisis de farmacocinética poblacional de los efectos de la co-medicación avala el uso de antiácidos y supresores del ácido gástrico sin ajuste de dosis de etexilato de dabigatrán en pacientes (BISTRO II) y mostró la ausencia de interacciones farmacológicas de dabigatrán con los fármacos más comúnmente usados en la población de estudio: opioides, diuréticos, paracetamol, fármacos antiinflamatorios no esteroideos, inhibidores de la ciclo-oxigenasa 2, inhibidores de la hidroximetilglutaril-coenzima A (HMG-CoA) reductasa, fármacos reductores de colesterol/triglicéridos no estatinas, antagonistas de la angiotensina II, inhibidores de la enzima convertidora de angiotensina, antagonistas del adrenorreceptor B, bloqueadores de los canales de Ca dihidropiridínicos, propulsores, derivados de las benzodiacepinas y fármacos conocidos por inhibir el transportador de la glicoproteína P (P-gp), y sustratos de la P-gp. Farmacodinamia: el etexilato de dabigatrán es una molécula pequeña que no presenta ninguna actividad farmacológica, debido a que actúa como prodroga. Después de la administración oral etexilato de dabigatrán es rápidamente absorbido y transformado a dabigatrán por medio de la hidrólisis catalizada por estereasas en plasma y en el hígado. Dabigatrán es un potente, competitivo, inhibidor directo reversible de la trombina y es el principal principio activo en el plasma. Puesto que la trombina (proteasa de serina) permite la conversión del fibrinógeno en fibrina en la cascada de coagulación, su inhibición previene el desarrollo del trombo. Dabigatrán también inhibe la trombina libre, la unión de la fibrina a la trombina y la agregación plaquetaria inducida por la trombina. Los estudios in vivo y ex vivo en animales han demostrado la eficacia antitrombótica y la actividad anticoagulante de dabigatrán después de su administración intravenosa y de etexilato de dabigatrán después de su administración oral en varios modelos de trombosis en animales. Etexilato de dabigatrán prolonga el tiempo de tromboplastina parcial activada (TTPa). En pacientes que están sangrando, las pruebas de TTPa pueden ser útiles para determinar un exceso de actividad anticoagulante, a pesar de que TTPa es menos sensible a la actividad de dabigatrán a niveles supraterapéuticos. Si están disponibles, el tiempo de trombina (TT) y el tiempo de coagulación de ecarina (ECT) pueden ser pruebas más sensibles para evaluar los efectos anticoagulantes de dabigatrán. El tiempo de protrombina (INR) se prolonga con dabigatrán, pero es menos sensible que el TT y ECT. Ensayos clínicos en profilaxis de ETV luego de cirugía mayor de reemplazo articular: en dos ensayos, aleatorizados, de grupos paralelos, doble ciego, confirmatorios de dosis, pacientes sometidos a cirugía electiva ortopédica mayor (uno para cirugía de reemplazo de rodilla y uno para cirugía de reemplazo de cadera) recibieron etexilato de dabigatrán 75 mg o 110 mg dentro de 1-4 horas de la cirugía, seguido de 150 o 220 mg diarios posteriormente, habiendo asegurado la hemostasia, o enoxaparina 40 mg en el día previo a la cirugía y luego diariamente. En el ensayo RE-MODEL (reemplazo de rodilla) el tratamiento fue de 6 a 10 días y el ensayo RE-NOVATE (reemplazo de cadera) de 28 a 35 días. Se trataron números totales de 2.076 pacientes (rodilla) y 3.494 (cadera) respectivamente. Los resultados del estudio de rodilla (RE-MODEL) con respecto al parámetro de evaluación primario, enfermedad tromboembólica venosa (ETV) total incluyendo asintomático, más morbilidad de cualquier causa, mostraron que el efecto antitrombótico de ambas dosis de etexilato de dabigatrán fue estadísticamente no inferior al de enoxaparina. Asimismo, la ETV total incluyendo asintomática, y la mortalidad de cualquier causa constituyeron el parámetro de evaluación primario para el estudio de cadera (RE-NOVATE). Nuevamente etexilato de dabigatrán con ambas dosis diarias fue estadísticamente no inferior a enoxaparina a 40 mg diarios. En un tercer estudio de asignación al azar, con grupo paralelo, doble ciego (RE-MOBILIZE), pacientes a quienes se les practicó cirugía de reemplazo total de rodilla recibieron etexilato de dabigatrán 75 o 110 mg en las 6 a 12 horas después de la cirugía, continuando con 150 mg y 220 mg diarios a partir de ahí. La duración del tratamiento fue de 12-15 días. En total 2.615 pacientes fueron asignados al azar y 2.596 fueron tratados. La dosis de enoxaparina fue de 30 mg bid, de acuerdo con la dosis aprobada en Estados Unidos. En el estudio RE-MOBILIZE no se estableció la no inferioridad. No hubo diferencias estadísticamente significativas en cuanto a sangrados entre los comparadores. También se evaluó un estudio de asignación al azar, con grupo paralelo, doble ciego, con fase placebo-controlada en pacientes japoneses, donde se administró etexilato de dabigatrán a dosis de 110 mg, 150 mg y 220 mg al día siguiente de la cirugía electiva de reemplazo total de rodilla. El estudio japonés mostró una clara relación dosis respuesta para la eficacia de dabigatrán y un perfil de sangrado similar al placebo. En RE-MODEL y RE-NOVATE la asignación al azar fue realizada antes de la cirugía, y en los estudios RE-MOBILIZE y el estudio japonés controlado con placebo, la asignación al azar fue hecha después de la cirugía. Esto es para notarlo especialmente en la seguridad de estos estudios. Por esta razón, en la tabla 2, los estudios se agruparon en estudios asignados al azar pre y post cirugía. Los datos del parámetro de ETV mayor y la mortalidad relacionada a ETV y los parámetros de sangrado mayor adjudicados se muestran en las tablas a continuación: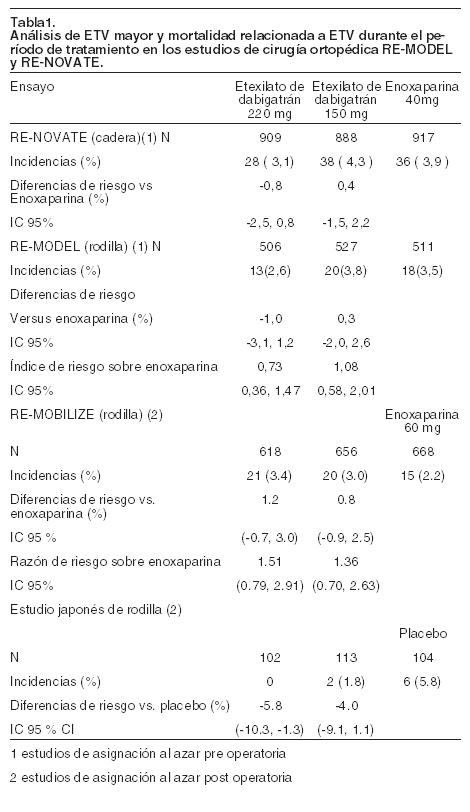
Contraindicaciones: Hipersensibilidad conocida a dabigatrán o etexilato de dabigatrán o a alguno de los excipientes de la fórmula. Pacientes con insuficiencia renal severa (CrCl < 30 ml/min). Manifestaciones hemorrágicas, pacientes con diátesis hemorrágica, o pacientes con alteración espontánea o farmacológica de la hemostasia. Lesiones orgánicas con riesgo significativo de sangrado, incluyendo accidente vascular cerebral hemorrágico ocurrido durante los últimos 6 meses. Pacientes con presencia de catéter espinal o epidural o durante la primera hora posterior al retiro del mismo.
Precauciones generales: Insuficiencia hepática: pacientes con insuficiencia hepática moderada y severa (Child-Pugh B o C) o enfermedad hepática que se espera que tenga algún impacto en la sobrevida o con elevación de las enzimas hepáticas > 2 ULN, los cuales fueron excluidos en los estudios clínicos. Por lo tanto, el uso de etexilato de dabigatrán no se recomienda en esta población. Riesgo hemorrágico: los siguientes tratamientos no deberán ser administrados concomitantemente con etexilato de dabigatrán: heparinas no fraccionadas y derivados de heparina, heparinas de bajo peso molecular, fondaparinux, desirudin, agentes trombolíticos, antagonistas del receptor GPIIb/IIIa, clopidogrel, ticlopidina, dextran, sulfinpirazona y antagonistas de la vitamina K. Deberá observarse que la heparina no fraccionada puede ser administrada en las dosis necesarias para mantener un catéter venoso o un catéter arterial central permeable. Se ha observado que el ácido acetilsalicílico a dosis de 75 a 325 mg incrementa el riesgo de sangrado cuando se administra concomitantemente con etexilato de dabigatrán a dosis por arriba de las recomendadas para la prevención de eventos de trombosis venosa. Las dosis recomendadas de etexilato de dabigatrán no han mostrado evidencia de incrementar el riesgo de sangrado atribuible a dabigatrán en pacientes que están recibiendo dosis bajas de ácido acetilsalicílico para la prevención de eventos cardiovasculares. Sin embargo, la información es limitada por lo que la coadministración de bajas dosis de ácido acetilsalicílico y etexilato de dabigatrán deben ser acompañadas por una observación clínica de sangrado. Se requiere una observación minuciosa (buscando signos de sangrado o anemia) en el seguimiento de situaciones que puedan incrementar el riesgo hemorrágico. Biopsia reciente o trauma mayor. Pacientes que están recibiendo tratamientos que incrementan el riesgo hemorrágico. La asociación de etexilato de dabigatrán con tratamientos que actúan en la hemostasia o coagulación puede incrementar el riesgo hemorrágico. Se ha observado que la administración de AINEs para analgesia perioperatoria por corto tiempo no está asociada con el incremento en el riesgo de sangrado cuando se administra en conjunto con etexilato de dabigatrán. Existe evidencia limitada con respecto al uso regular de medicación con AINEs con vida media menor a 12 horas durante el tratamiento con etexilato de dabigatrán, sin embargo no se ha observado un riesgo adicional de sangrado. El uso oral de un inhibidor potente e P-gp (ej., verapamil) en forma concomitante con etexilato de dabigatrán puede elevar las concentraciones plasmáticas de dabigatrán, resultando en un incremento en el riesgo de sangrado. Se debe evitar iniciar el tratamiento con verapamil en pacientes que han tenido una cirugía ortopédica mayor que estén siendo tratados con etexilato de dabigatrán. El inicio simultáneo de tratamiento con etexilato de dabigatrán y verapamil se debe evitar. Endocarditis bacteriana. Insuficiencia renal: estudios farmacocinéticos demostraron un incremento en la exposición al medicamento en pacientes con función renal reducida incluyendo el declive de la función renal relacionado con la edad. Se recomienda reducir la dosis a 150 mg al día en pacientes con insuficiencia renal moderada (50-30ml/min). Dabigatrán está contraindicado en casos de insuficiencia renal severa (CrCl < 30 ml/min). Los pacientes en quienes se descubra falla renal aguda deberán descontinuar dabigatrán. Anestesia espinal/anestesia epidural/punción lumbar: el riesgo de hematoma espinal o epidural puede incrementarse en casos de punción traumática o punciones repetidas y por el uso prolongado de catéteres epidurales postoperatorios. Después de remover un catéter, debe transcurrir un intervalo de por lo menos 1 hora antes de la administración de la primera dosis de dabigatrán. Estos pacientes requieren frecuente observación de los signos y síntomas neurológicos. El producto contiene el excipiente amarillo amanecer, que puede causar reacciones alérgicas.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: no existen datos disponibles de exposición en pacientes embarazadas. El riesgo en humanos con esta condición es desconocido. Estudios en animales en reproducción no mostraron efectos adversos en la fertilidad o en el desarrollo postnatal del neonato. Las mujeres con potencial de fertilidad deben evitar el embarazo durante el tratamiento con dabigatrán y en caso de embarazo las mujeres no deberán ser tratadas con dabigatrán a menos que el beneficio previsto sea mucho mayor que el riesgo. Lactancia: no existen datos clínicos disponibles. Como una precaución, deberá ser suspendida la lactancia cuando se administre dabigatrán.
Reacciones secundarias y adversas: Un total de 10.596 pacientes fueron tratados en 5 estudios controlados de prevención de trombosis venosa con por lo menos una dosis del medicamento en estudio. De ellos 5.674 fueron tratados con 150 o 220 mg de dabigatrán diariamente, mientras que 522 recibieron dosis menores de 150 mg diarios y 1.168 recibieron dosis superiores a 220 mg al día. Las reacciones adversas que pueden certeramente ser atribuidas a dabigatrán, y que ocurrieron con similar frecuencia con enoxaparina, son aquellas relacionadas con sangrado o signos de sangrado ej., anemia, sangrado a través de una herida. La definición de eventos de sangrado mayor siguió los criterios de ISTH (Sociedad Internacional de Trombosis y Hemostasia, por sus siglas en inglés) y las guías de la EMEA. De acuerdo al sistema de codificación del MedDRA los eventos de sangrado están distribuidos por órganos y sistemas; un resumen descriptivo de sangrado mayor y cualquier sangrado se encuentra en la tabla 1. Aunque la frecuencia de los sangrados mayores o severos es rara en los estudios clínicos, éstos pueden ocurrir, y, de acuerdo a su localización, pueden causar discapacidad, amenazar la vida o tener consecuencias fatales. La tabla 2 muestra el número (%) de pacientes que experimentaron diferentes grados de eventos de sangrado durante el período de tratamiento en la prevención de trombosis venosa de acuerdo con la dosis.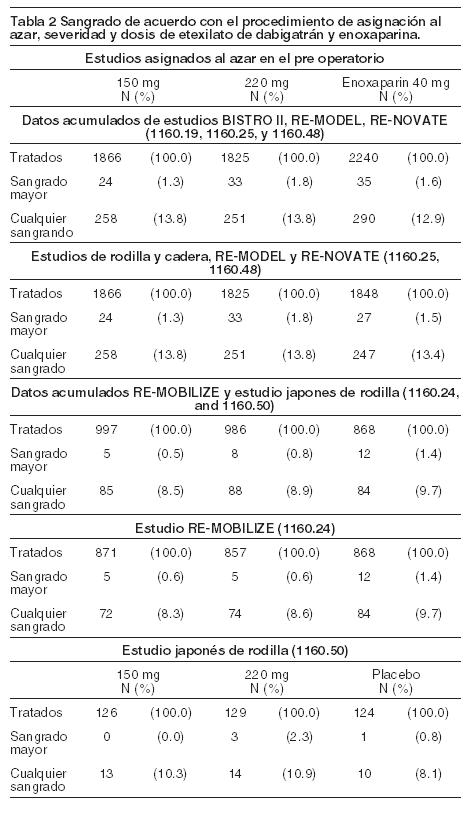
Las tasas de sangrado en general fueron similares entre los grupos de tratamiento y no estadísticamente diferentes. Las reacciones adversas fueron clasificadas por aparatos y sistemas y en los términos elegidos se comunicaron de cualquier grupo de tratamiento de todos los pacientes tratados en los estudios controlados de la prevención de trombosis venosa son enlistados a continuación: trastornos del sistema linfático y sanguíneo: anemia, trombocitopenia. Trastornos vasculares: hematoma, hemorragia, hemorragia de la herida. Trastornos respiratorios, torácicos y mediastinales: epistaxis. Trastornos gastrointestinales: sangrado gastrointestinal, sangrado rectal, sangrado hemorroidal. Trastornos hepatobiliares: función hepática anormal. Trastornos de la piel y tejido subcutáneo: sangrado en la piel. Trastornos musculoesqueléticos, óseos y del tejido conectivo: hemartrosis. Trastornos renales y urinarios: hematuria. Trastornos generales y condiciones del sitio de administración: sangrado del sitio de inyección, sangrado del sitio del catéter, secreción hemorrágica. Lesiones, envenenamiento y complicaciones del procedimiento: secreción de la herida, hematoma post-procedimiento, anemia post-cirugía, hematoma traumático, secreción en el sitio del procedimiento, sangrado en el sitio de incisión. Procedimientos quirúrgicos y médicos: drenaje post-procedimiento, drenaje de la herida. Las incidencias observadas de las reacciones adversas de etexilato de dabigatrán estaban en el rango de las de enoxaparina. Para el producto etiquetado con presentación de 150 mg: las frecuencias de las reacciones presentadas para el producto aplican para todos los pacientes y no sólo en pacientes con deterioro de la función renal. Aunque su frecuencia ha sido rara en los estudios clínicos, el sangrado mayor o severo puede ocurrir y, independientemente de la localización, puede llevar a la discapacidad, amenaza a la vida o aun a desenlaces fatales.
Interacciones medicamentosas y de otro género: El uso concomitante de dabigatrán con tratamientos que actúan en la hemostasia o coagulación incluyendo antagonistas de la vitamina K, pueden incrementar importantemente el riesgo de sangrado. Ver Precauciones generales. Etexilato de dabigatrán y dabigatrán no se metabolizan por el sistema de citocromo P450 y no tienen efectos in vitro en las enzimas del citocromo P450 en humanos. Por lo tanto, no se esperan interacciones droga-droga relacionadas con etexilato de dabigatrán y dabigatrán. Amiodarona: cuando etexilato de dabigatrán fue coadministrado con una dosis única de 600 mg de amiodarona, el grado y el índice de absorción de amiodarona y de su metabolito activo DEA permanecieron sin cambios. El área bajo la curva (AUC) y la Cmáx de dabigatrán se incrementaron en un 60% y 50% respectivamente. Verapamil: cuando se coadministró etexilato de dabigatrán con verapamil oral, la Cmáx y ABC de dabigatrán se incrementaron dependiendo del momento de la administración de verapamil y de su formulación. La mayor elevación de la exposición de dabigatrán se observó con la primera dosis de una formulación de liberación inmediata de verapamil, administrado una hora antes de la toma de etexilato de dabigatrán (incremento de Cmáx cerca del 180% y ABC cerca del 150%). El efecto disminuyó progresivamente con la administración de una formulación de liberación prolongada (incremento de Cmáx de cerca del 90% y de ABC de alrededor del 70%) o de la administración de múltiples dosis de verapamil (incremento de Cmáx de cerca del 60% y del ABC de alrededor del 50%). Esto se puede explicar por la inducción e P-gp en el intestino por tratamiento crónico con verapamil. No hubo una interacción significativa cuando verapamil se dio 2 horas después de etexilato de dabigatrán (incremento del Cmáx de alrededor del 10% y del ABC de alrededor del 20%). Esto se explica por la absorción completa de dabigatrán después de 2 horas. (Ver Dosis y vía de administración). No hay datos disponibles para la aplicación parenteral de verapamil; basándose en el mecanismo de interacción, no se espera ninguna interacción significativa. Claritromicina: cuando claritromicina se administró a una dosis de 500 mg bid junto con etexilato de dabigatrán no hubo interacciones relevantes farmacocinéticas (incremento de Cmáx alrededor del 19% y de ABC alrededor del 15%). Atorvastatina: cuando etexilato de dabigatrán fue coadministrado con atorvastatina, la exposición a atorvastatina, metabolitos de atorvastatina y de dabigatrán no mostraron cambios indicando carencia de interacción. Diclofenaco: cuando etexilato de dabigatrán fue coadministrado con diclofenaco, la farmacocinética de ambos fármacos permanecieron sin cambios indicando una ausencia de interacción entre etexilato de dabigatrán y diclofenaco. La coadministración de AINEs por corto tiempo para analgesia perioperatoria no ha sido asociada con el incremento en el riesgo de sangrado. Existe limitada experiencia con respecto a la seguridad de de etexilato de dabigatrán cuando es coadministrado a largo plazo. Digoxina: cuando etexilato de dabigatrán fue coadministrado con digoxina no se observó interacción farmacocinética. Pantoprazol: cuando etexilato de dabigatrán fue coadministrado con pantoprazol, se observó una disminución en el tiempo de la concentración plasmática del área bajo la curva aproximadamente del 30%. Pantoprazol y otros inhibidores de bomba de protones fueron coadministrados con etexilato de dabigatrán en estudios clínicos sin observarse efectos en el sangrado ó la eficacia. Ranitidina: la administración de ranitidina junto con etexilato de dabigatrán, no tuvo ningún efecto clínicamente relevante en el grado de absorción de dabigatrán.
Alteraciones en los resultados de pruebas de laboratorio: Han sido observados algunos cambios no significativos en los niveles de las enzimas hepáticas (ver Reacciones adversas).
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Los estudios de teratogenicidad fueron realizados con dosis mayores de 200 mg/kg en ratas y conejos. Fue observado un efecto leve sobre la morfogénesis de los fetos de ratas con dosis de 200 mg/kg. No fueron observados efectos teratogénicos en conejos. En estudios de fertilidad en ratas, no fueron observados efectos toxicológicos notables. Los estudios in vitro e in vivo no revelaron evidencia de potencial mutagénico.
Dosis y vía de administración: Prevención de tromboembolismo venoso en pacientes con cirugía ortopédica mayor: la dosis recomendada de dabigatrán es de 220 mg una vez al día (2 cápsulas de 110 mg). Pacientes con insuficiencia renal moderada tienen un incremento de riesgo de sangrado. Para estos pacientes la dosis recomendada de dabigatrán es de 150 mg al día, tomando dos cápsulas de 75 mg. Prevención de tromboembolismo venoso seguido de cirugía de reemplazo de rodilla: el tratamiento con dabigatrán debe ser iniciado por vía oral en un plazo de 1 a 4 horas terminada la cirugía con una sola cápsula (110 mg) y continuar con 2 cápsulas una vez al día para completar un total de 10 días. Si la hemostasia no es segura, deberá retrasarse el inicio del tratamiento. Si el tratamiento no es iniciado el día de la cirugía, entonces el tratamiento deberá ser iniciado con 2 cápsulas al día. Prevención de tromboembolismo venoso seguido de cirugía de reemplazo de cadera: el tratamiento con dabigatrán debe ser iniciado por vía oral en un plazo de 1 a 4 horas terminada la cirugía con una sola cápsula (110 mg) y continuar con 2 cápsulas una vez al día para completar un total de 28 a 35 días. Si la hemostasia no es segura, deberá retrasarse el inicio del tratamiento. Si el tratamiento no es iniciado el día de la cirugía, entonces el tratamiento deberá ser iniciado con 2 cápsulas al día. Dabigatrán deberá tomarse con agua, con o sin alimentos. Insuficiencia hepática: pacientes con insuficiencia hepática moderada y severa (clasificación Child-Pugh B y C) o enfermedad hepática que se espera tenga impacto en la sobrevida o con elevación de las enzimas hepáticas > 2 veces por arriba de los límites normales, los cuales fueron excluidos de los estudios clínicos. Por lo tanto, el uso del dabigatrán no se recomienda en esta población. Insuficiencia renal: posterior a la aplicación intravenosa, el 85% del dabigatrán en plasma es depurado a nivel renal. Los pacientes con insuficiencia renal moderada (depuración de creatinina 30-50 ml/min) pueden estar en riesgo alto de sangrado. La dosificación deberá ser reducida a 150 mg de dabigatrán diariamente en pacientes con insuficiencia renal moderada. La depuración de creatinina puede ser estimada usando la fórmula de Cockroft-Gault como sigue:
No existen datos que soporten el uso en pacientes con insuficiencia renal severa depuración de creatinina ( < 30ml/min); no se recomienda el tratamiento con dabigatrán en esta población. Dabigatrán puede ser dializado, no existe experiencia clínica que demuestre la utilidad de este punto en estudios clínicos. Ancianos: los estudios clínicos han sido conducidos en una población de pacientes con una edad promedio > 65 años. En general los pacientes deben ser tratados con 220 mg de dabigatrán al día. Los estudios farmacocinéticos en personas ancianas demuestran un incremento en la exposición al medicamento en aquellos pacientes con disminución de la función renal relacionada con la edad. Ver Dosis y vía de administración en Insuficiencia renal. Niños: dabigatrán no ha sido investigado en pacientes menores de 18 años de edad. No se recomienda el tratamiento con dabigatrán en niños. Peso: en un estudio con un modelo farmacocinético mostró que los pacientes con un peso corporal de 120 kg tienen cerca de 20% menor exposición al fármaco y que los pacientes con un peso corporal de 48 kg tienen cerca del 25% mayor exposición al producto comparado con pacientes con un peso corporal promedio (BISTRO II). Sin embargo no existió diferencia en la eficacia y los rangos de sangrado, por lo que no es necesario realizar un ajuste de dosis. Uso concomitante de etexilato de dabigatrán con amiodarona o verapamil: la dosis de etexilato de dabigatrán debe reducirse a 150 mg al día en pacientes que reciban concomitantemente etexilato de dabigatrán con amiodarona o verapamil. Rotación del tratamiento con etexilato de dabigatrán a anticoagulantes parenterales: esperar 24 horas después de la última dosis antes de realizar el cambio de dabigatrán a anticoagulante parenteral. Rotación del tratamiento con un anticoagulante parenteral a etexilato de dabigatrán: no existen datos disponibles, por lo que no se recomienda comenzar con la administración de dabigatrán antes de que tuviera que aplicarse la siguiente dosis programada de anticoagulante parenteral.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: No existe antídoto para dabigatrán. Las dosis de dabigatrán más allá de la exposición recomendada representa un riesgo importante de sangrado. En caso de complicaciones hemorrágicas, el tratamiento debe ser suspendido y la fuente de sangrado deberá ser investigada. Debido a que el dabigatrán es excretado predominantemente por vía renal deberá ser mantenida una adecuada diuresis. Deberá ser considerado el inicio de tratamiento apropiado, ej., hemostasia quirúrgica o de transfusión de plasma fresco congelado. Dabigatrán puede ser dializado, no existe experiencia clínica que demuestre la utilidad de este punto en estudios clínicos. Casos reportados de pacientes con eventos hemorrágicos con dosis superiores a 300 mg diarios: Existen estudios en voluntarios sanos donde etexilato de dabigatrán fue bien tolerado sin evidencia desangrado con dosis desde 10 a 400 mg, dados hasta 3 veces al día durante 7 días. Ocurrieron equímosis en los sitios de venopunción y sangrado gingival a dosis de 400 mg, tres veces al día. En el estudio multicéntrico BISTRO I, cuyo objetivo era determinar el rango terapéutico de seguridad de etexilato de dabigatrán en pacientes con reemplazo total de cadera (n=314), hubo grupos de pacientes a los que se les administraron dosis de 200 mg y 300 mg dos veces al día, cuatro a 8 horas después de la cirugía, durante 6 a 10 días. El objetivo primario de seguridad fue sangrado. Dentro de los resultados no hubo sangrados mayores en ninguna de las dosis, sólo dos pacientes con la mayor dosis (300 mg dos veces al día) presentaron sangrados de múltiples sitios y se asoció a disminución de la función renal y parámetros farmacodinámicos prolongados. En el estudio BISTRO II (n=1.464), etexilato de dabigatrán se comparó con enoxaparina para la prevención de enfermedad tromboembólica después de reemplazo de cadera o rodilla. Se utilizaron dosis de dabigatrán desde 50 mg dos veces al día hasta 225 mg dos veces al día. Los resultados de seguridad mostraron que los episodios de sangrado mayor eran significativamente mayores en los pacientes con las dosis de 150 mg dos veces al día, 225 mg dos veces al día y 300 mg una vez al día, que en los de dosis 50 mg dos veces al día. Comparado con enoxaparina 40 mg al día, en las dosis 150 mg dos veces al día, 225 mg dos veces al día y 300 mg una vez al día hubo una tendencia a más sangrados mayores, sin ser estadísticamente significativa. Los sangrados que requirieron suspensión del tratamiento no fueron estadísticamente significativos en ninguna de las dosis de etexilato de dabigatrán, comparado con enoxaparina.
Presentación(es): Caja con 10, 30 o 60 cápsulas de 75 o 110 mg. Caja con frasco con 10, 30 o 60 cápsulas de 75 o 110 mg.
Recomendaciones sobre almacenamiento: Blíster: almacenar en el envase original para proteger de la humedad. Consérvese a temperatura ambiente a no más de 25°C. Frasco: una vez abierto, el producto debe utilizarse dentro de los siguientes 30 días. Almacenar en el envase original para proteger de la humedad. Consérvese el frasco bien tapado a temperatura ambiente a no más de 25°C.
Leyendas de protección: Literatura exclusiva para médicos. Dosis: la que el médico señale. Su venta requiere receta médica. No se deje al alcance de los niños. No se use en embarazo ni en la lactancia. Léase instructivo anexo. El frasco contiene un desecante. No ingerible, consérvese dentro del envase.
Nombre y domicilio del laboratorio: Hecho en Alemania por: Boehringer Ingelheim Pharma GmbH & Co. KG, Birkendorfer Strasse 65 D-88397 Biberach, Alemania ó Boehringer Ingelheim Pharma GmbH & Co. KG, Binger Strasse 173 55216 Ingelheim am Rhein. Acondicionado en Alemania por: Boehringer Ingelheim Pharma GmbH & Co. KG, Binger Strasse 173 55216 Ingelheim am Rhein. Distribuido por: Boehringer Ingelheim Promeco S.A. de C.V., Calle del Maíz No. 49, Barrio Xaltocan, Xochimilco. 16090 México, D.F.
Número de registro del medicamento: 358M2008 SSA IV.
Clave de IPPA: 093300C0017607