PROGRAF
JANSSEN-CILAG
Denominación genérica: Tacrolimus
Forma farmacéutica y formulación: PROGRAF® cápsulas: Cada CÁPSULA contiene: Tacrolimus monohidratado equivalente a 0.5 mg de tacrolimus, Excipiente, cbp 1 cápsula. Tacrolimus monohidratado equivalente a 1 mg de tacrolimus Excipiente, cbp 1 cápsula. Tacrolimus monohidratado equivalente a 5 mg de tacrolimus, Excipiente, cbp 1 cápsula.
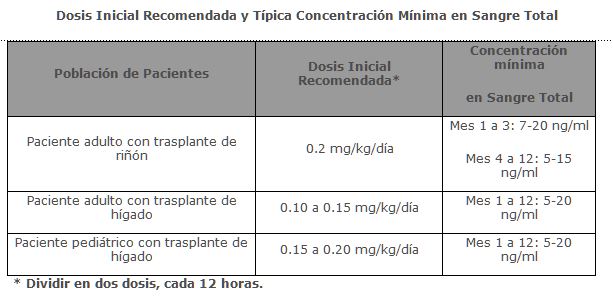
Indicaciones terapéuticas: PROGRAF® está indicado para la profilaxis del rechazo de órganos en pacientes que recibieron trasplantes alogénicos de riñón o hígado. Se recomienda que PROGRAF® sea usado concomitantemente con corticoesteroides adrenérgicos. Debido al riesgo de anafilaxis PROGRAF® Solución inyectable debe usarse sólo en pacientes que no puedan ingerir cápsulas de PROGRAF®.
Farmacocinética y farmacodinamia: Tacrolimus es el ingrediente activo de PROGRAF®. Tacrolimus es un macrólido inmunosupresor producido por Streptomyces tsukubaensis. Se presenta en cristales blancos o polvo cristalino. Es prácticamente insoluble en agua, ligeramente soluble en etanol y muy soluble en metanol y cloroformo. Mecanismo de acción: Tacrolimus prolonga la supervivencia del huésped y del órgano trasplantado en modelos animales receptores de trasplante de hígado, riñón, corazón, médula ósea, intestino delgado y páncreas, pulmón y tráquea, piel, córnea y extremidades. Tacrolimus ha demostrado que suprime en cierta medida la inmunidad humoral en animales, y en gran medida las reacciones mediadas por células tales como rechazo alogénico, hipersensibilidad retardada, artritis inducida por colágeno, encefalomielitis alérgica experimental y la enfermedad de injerto contra huésped. Tacrolimus inhibe la activación de linfocitos-T, aunque el mecanismo de acción exacto no es conocido. Las evidencias experimentales sugieren que el Tacrolimus se une a una proteína intracelular, FKBP-12. Se forma entonces un complejo de Tacrolimus-FKBP-12, calcio, calmodulina y calcineurina con lo que la actividad de la fosfatasa de calcineurina se inhibe. Este efecto puede prevenir la defosforilación y translocación de un factor nuclear de las células-T activadas (NF-AT), un componente nuclear que se considera inicia la transcripción del gen para la formación de linfocinas (tales como interleucina-2 e interferon gama). El resultado total es la inhibición de la activación del linfocito-T (por ejemplo, inmunosupresión). Farmacocinética: La actividad de Tacrolimus se debe principalmente al fármaco padre. Los parámetros farmacocinéticos han sido determinados después de la administración intravenosa (IV) y oral (VO) en voluntarios sanos; y en pacientes con trasplante de hígado y pacientes con trasplante de riñón.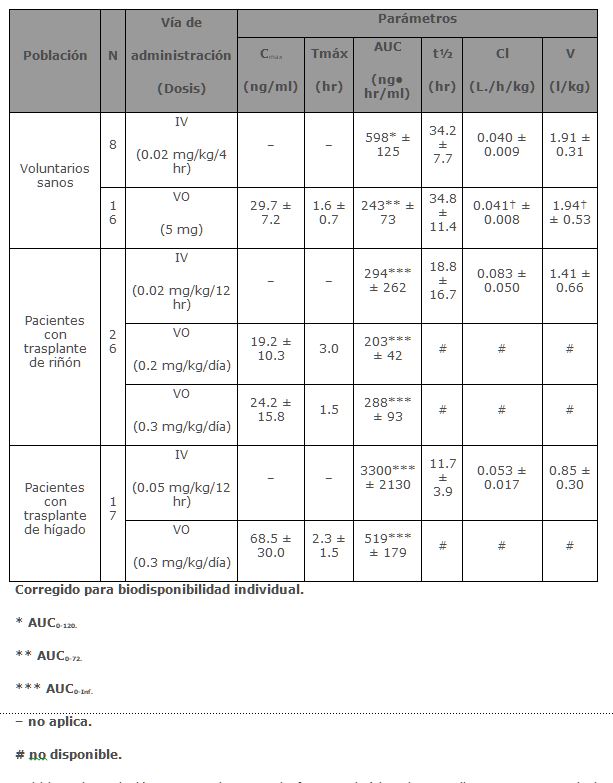
Debido a la variación entre sujetos en la farmacocinética de Tacrolimus, es necesaria la individualización del esquema de dosis para una terapia óptima. Los datos farmacocinéticos indican que la concentración en sangre total comparada con la concentración en plasma es el compartimento muestra más apropiado para describir la farmacocinética de Tacrolimus. Absorción: La absorción de Tacrolimus a partir del tracto gastrointestinal después de su administración oral es incompleta y variable. La biodisponibilidad absoluta de Tacrolimus fue 17 ± 10% en pacientes adultos con trasplante de riñón (N = 26), 22 ± 6% en pacientes adultos con trasplante de hígado (N = 17) y 18 ± 5% en voluntarios sanos (N = 16). Un estudio de dosis única realizado en 32 voluntarios sanos estableció la bioequivalencia de las cápsulas de 1 mg y 5 mg. Otro estudio de dosis única en 32 voluntarios sanos estableció la bioequivalencia de las cápsulas de 0.5 mg y 1 mg. La concentración máxima de Tacrolimus en sangre (Cmax) y el área bajo la curva (AUC por sus siglas en inglés) parece que se incrementan en una forma proporcional a la dosis en 18 voluntarios sanos en ayuno que recibieron una dosis única de 3, 7 y 10 mg. En 18 pacientes con trasplante de riñón, Tacrolimus tuvo concentraciones de 3 a 30 ng/ml medidas a las 10-12 horas post-dosis (Cmin) correlacionando bien con el AUC (Coeficiente de correlación 0.93). En 24 pacientes con trasplante de hígado con un intervalo de concentración de 10 a 60 ng/ml, el coeficiente de correlación fue 0.94. Efecto de los alimentos: La tasa y extensión de la absorción de Tacrolimus fue mayor en condiciones de ayuno. La presencia y composición de los alimentos disminuyó la velocidad y área de la absorción de Tacrolimus cuando se administró en 15 voluntarios sanos. El efecto fue más pronunciado con alimentos altos en grasa (848 Kcal, 46% grasa), con lo que los promedios de AUC y Cmax disminuyeron 37% y 77% respectivamente; el Tmax aumentó 5 veces. Una comida alta en carbohidratos (668 kcal, 85% carbohidratos) disminuyó los promedios de el AUC y la Cmax en un 28% y 65% respectivamente. En voluntarios sanos (N = 16) el tiempo de la comida también afectó la biodisponibilidad de Tacrolimus. Cuando se administró inmediatamente después de la comida, el promedio de la Cmax disminuyó 71% y el promedio del AUC se redujo 39% respecto a las condiciones de ayuno. En 11 pacientes con trasplante de hígado se administró PROGRAF® 15 minutos después de un desayuno alto en grasa (400 Kcal, 34% grasa), lo que produjo una disminución del AUC (27 ± 18%) y de la Cmax (50 ± 19%) comparado con las condiciones de ayuno. Distribución: La unión de Tacrolimus a proteínas plasmáticas es aproximadamente del 99% y es independiente de la concentración en un intervalo de 5-50 ng/ml. Tacrolimus se une principalmente a la albúmina y a la alfa-1-ácido glicoproteína, y tiene un nivel alto de asociación con eritrocitos. La distribución de Tacrolimus entre sangre total y plasma depende de varios factores, tales como hematócrito, la temperatura en el momento de la separación del plasma, concentración del fármaco y concentración de proteínas plasmáticas. En un estudio en EUA la proporción de la concentración en sangre total sobre la concentración en plasma tuvo un promedio de 35 (intervalo de 12 a 67). Metabolismo: Tacrolimus es ampliamente metabolizado por un sistema de oxidasa de función mixta, principalmente el sistema citocromo P-450 (CYP3A). Se ha propuesto una ruta metabólica que conduce a la formación de 8 posibles metabolitos. Los principales mecanismos de biotransformación que fueron detectados in vitro son la desmetilación y la hidroxilación. El principal metabolito identificado en incubación con microsomas de hígado humano es el Tacrolimus 13-desmetilado. En un estudio in vitro se reportó que el metabolito 31-desmetilado tiene la misma actividad que el Tacrolimus. Excreción: El promedio de depuración después de la administración IV de Tacrolimus es 0.040, 0.083 y 0.053 L/hr/kg en voluntarios sanos, pacientes adultos con trasplante de riñón y pacientes adultos con trasplante de hígado respectivamente. En el hombre, menos del 1% de la dosis administrada se excretó sin cambios en la orina. En un estudio de balance de masas con Tacrolimus marcado radioactivamente administrado por vía IV a 6 voluntarios sanos, el promedio de recuperación de la sustancia marcada radioactivamente fue de 77.8 ± 12.7%. La eliminación fecal fue de 92.4 ± 1.0% y la vida media de eliminación con base en la radioactividad fue de 48.1 ± 15.9 horas mientras que con base en las concentraciones de Tacrolimus fue de 43.5 ± 11.6 horas. El promedio de depuración de la sustancia radiomarcada fue de 0.029 ± 0.015 L/hr/kg y la depuración de Tacrolimus fue de 0.029 ± 0.009 L/hr/kg. Cuando se administró por vía oral, el promedio de recuperación de la sustancia marcada radioactivamente fue de 94.9 ± 30.7%. La eliminación fecal fue de 92.6 ± 30.7%, la eliminación urinaria fue de 2.3 ± 1.1% y la vida media de eliminación con base en la radioactividad fue de 31.9 ± 10.5 horas mientras que con base en las concentraciones de Tacrolimus fue de 48.4 ± 12.3 horas. El promedio de depuración de la sustancia radiomarcada fue de 0.226 ± 0.116 L/hr/kg y la depuración de Tacrolimus fue de 0.172 ±0.088 L/hr/kg. Poblaciones especiales: Pediátrica: La farmacocinética de Tacrolimus se ha estudiado en pacientes con trasplante de hígado de 0.7 a 13.2 años de edad. Después de la administración IV a 12 pacientes pediátricos de 0.037 mg/kg/día los promedios de vida media, volumen de distribución y depuración fueron de 11.5 ± 3.8 horas, 2.6 ± 2.1 L/kg y 0.138 ± 0.071 L/hr/kg respectivamente. Después de la administración oral a 9 pacientes los promedios de AUC y Cmax. fueron 337 ± 167 ng• hr/ml y 43.4 ± 27.9 ng/ml respectivamente. La biodisponibilidad absoluta fue 31 ± 21%. Estas concentraciones en sangre total en 31 pacientes menores de 12 años, demuestran que los pacientes pediátricos requieren dosis más altas que los adultos para alcanzar concentraciones similares de Tacrolimus.Insuficiencia renal y hepática: El promedio de los parámetros farmacocinéticos para tacrolimus después de la administración de una dosis única a pacientes con daño hepático y renal están dados en la siguiente tabla.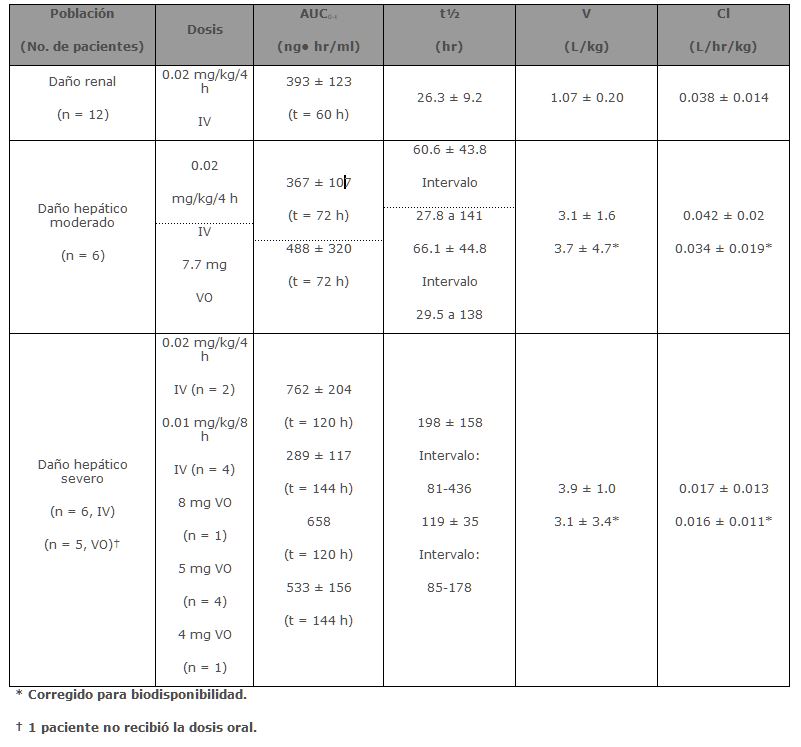
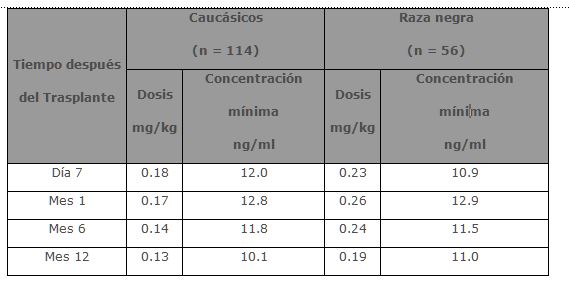
Insuficiencia renal: Los parámetros farmacéuticos para Tacrolimus después de una administración IV única se determinaron en 12 pacientes (7 sin diálisis y 5 con diálisis, con valores de creatinina sérica de 3.9 ± 1.6 y 12.0 ± 2.4 mg/dL, respectivamente) antes del trasplante de riñón. Los parámetros farmacocinéticos obtenidos fueron similares para ambos grupos. El promedio de depuración de Tacrolimus en pacientes con disfunción renal fue similar a la obtenida en voluntarios sanos. Insuficiencia hepática: La farmacocinética de Tacrolimus fue determinada en 6 pacientes con disfunción hepática moderada (promedio clasificación Pugh: 6.2) después de administraciones únicas IV y oral. El promedio de depuración de Tacrolimus en pacientes con disfunción hepática leve no difirió sustancialmente a la de voluntarios sanos. La farmacocinética de Tacrolimus se estudió en 6 pacientes con daño hepático severo (promedio clasificación Pugh: > 10). El promedio de depuración fue sustancialmente más bajo en pacientes con disfunción hepática severa, independientemente de la vía de administración. Raza: No se ha realizado un estudio formal para evaluar la disposición farmacocinética de Tacrolimus en pacientes trasplantados de raza negra. Sin embargo, una comparación retrospectiva de pacientes con trasplante renal, caucásicos y de raza negra indica que los pacientes de raza negra requieren dosis más altas de tacrolimus para obtener concentraciones similares. Género: No se ha realizado un estudio formal para evaluar el efecto del género en la farmacocinética de Tacrolimus, sin embargo, no hubo diferencia en la dosificación por género en el estudio de pacientes de trasplante de riñón. Una comparación retrospectiva de la farmacocinética en voluntarios sanos y en pacientes con trasplante de hígado y riñón indican que no hay diferencias basadas en el género. Farmacodinamia: Estudios clínicos: Trasplante de hígado: La seguridad y eficacia de la inmunosupresión con base en PROGRAF® después de un trasplante de hígado ortotópico se evaluó en dos estudios no-ciegos, aleatorizados, efectuados en varios centros. Los grupos de control activos fueron tratados con un régimen inmunosupresor a base de ciclosporina. Ambos estudios usaron concomitantemente corticoesteroides adrenérgicos como parte del régimen inmunosupresor. Estos estudios fueron diseñados para evaluar, como punto final primario, si los dos regímenes eran terapéuticamente equivalentes, tanto en la supervivencia de los pacientes como en la del órgano transplantado los siguientes 12 meses a la operación. Se encontró que el régimen inmunosupresor basado en PROGRAF® es equivalente a los regímenes inmunosupresores a base de ciclosporina. En el primer estudio se reclutaron 529 pacientes en 12 sitios clínicos de EUA. Antes de la cirugía 263 se asignaron aleatoriamente al programa inmunosupresor a base de PROGRAF® y 266 al régimen inmunosupresor a base de ciclosporina (CBIR, por sus siglas en inglés). En 10 de los 12 sitios se utilizó el mismo protocolo CBIR, mientras que los otros dos utilizaron diferentes protocolos de control. Este análisis excluyó a los pacientes con disfunción renal, falla hepática fulminante con encefalopatía fase IV y cánceres; los pacientes pediátricos (menores o iguales a 12 años) fueron admitidos. En el segundo estudio se reclutaron 545 pacientes en 8 sitios clínicos de Europa. Antes de la cirugía 270 se asignaron aleatoriamente al programa inmunosupresor a base de PROGRAF® y 275 a CBIR. En este estudio cada centro utilizó su protocolo CBIR estándar local, en el brazo de control activo. Este análisis excluyó a los pacientes pediátricos, pero admitió pacientes con disfunción renal, falla hepática fulminante con encefalopatía fase IV y cánceres diferentes a cáncer hepático primario con metástasis. La supervivencia a un año del paciente y del órgano trasplantado en los grupos con tratamiento a base de PROGRAF® fue equivalente a los grupos tratados con CBIR en ambos estudios. La supervivencia total del paciente a un año del trasplante (combinando grupos tratados con CBIR y a base de PROGRAF®) fue de 88% en el estudio de EUA y 78% en el estudio europeo. La supervivencia total del injerto a un año del trasplante (combinando grupos tratados con CBIR y con base en PROGRAF®) fue de 81% en el estudio de EUA y 73% en el estudio europeo. En ambos estudios el tiempo medio para pasar de la dosis IV a la dosis oral de PROGRAF® fue de dos días. Debido a la naturaleza del diseño del estudio, la comparación de diferencias en puntos finales secundarios tales como incidencia de rechazo agudo, rechazo refractario o el uso de OKT3 para rechazo resistente a esteroides, no puede llevarse a cabo. Trasplante de riñón: Se realizó un estudio, aleatorio, no ciego, en múltiples centros, Fase III para evaluar la inmunosupresión a base de PROGRAF® después de un trasplante de riñón. Se incluyeron 412 pacientes con trasplante de riñón en 19 clínicas de EUA. El estudio de la terapia se inició cuando la función renal se encontraba estable como lo indicaba la creatinina sérica ≤ 4 mg/dL (mediana de 4 días después del trasplante, intervalo de 1 a 14 días). Se excluyeron los pacientes menores a 6 años. Se asignaron aleatoriamente 205 pacientes al programa de inmunosupresión a base de PROGRAF® y 207 a CBIR. Todos los pacientes recibieron terapia de inducción profiláctica consistente en una preparación de anticuerpos antilinfocitos, corticoesteroides y azatioprina. La supervivencia total a un año del paciente y del órgano trasplantado fue 96.1% y 89.6% respectivamente y fueron equivalentes en ambos brazos del estudio. Debido a la naturaleza del diseño del estudio, la comparación de diferencias en puntos finales secundarios tales como incidencia de rechazo agudo, rechazo refractario o el uso de OKT3 para rechazo resistente a esteroides, no puede llevarse a cabo.
Contraindicaciones: PROGRAF® está contraindicado en pacientes con hipersensibilidad a Tacrolimus. PROGRAF® solución inyectable está contraindicado en pacientes con hipersensibilidad a HCO-60 (aceite de ricino polioxil 60 hidrogenado).
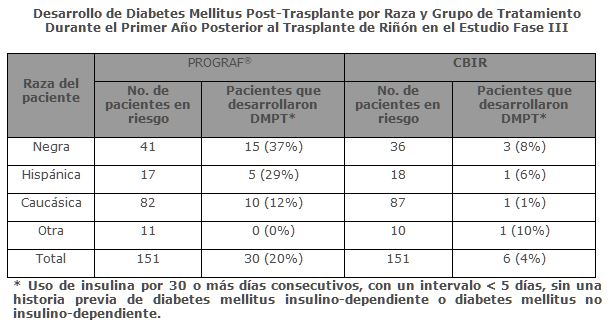
Precauciones generales: La inmunosupresión, puede dar como resultado, una susceptibilidad aumentada a la infección y el posible desarrollo de linfoma. Solamente los médicos experimentados en la terapia inmunosupresora y el manejo de los pacientes receptores de trasplantes de órganos deben prescribir PROGRAF®. Los pacientes que reciban el medicamento, deberán manejarse en instalaciones equipadas, con personal de laboratorio adecuado y recursos médicos de apoyo. El médico responsable de la terapia de mantenimiento deberá tener, como requisito, la información completa para el seguimiento del paciente. Generales: Se ha reportado diabetes mellitus insulino-dependiente post-trasplante (DMPT) en 20% de los pacientes con trasplante renal tratados con PROGRAF® sin historia de diabetes mellitus pretrasplante en el estudio Fase III. El tiempo promedio de inicio de la DMPT fue de 68 días. La insulino-dependencia fue reversible en 15% de estos pacientes a un año y en 50% a dos años post-trasplante. Los pacientes de raza negra e hispánica con trasplante renal tuvieron aumento en el riesgo de desarrollar DMPT.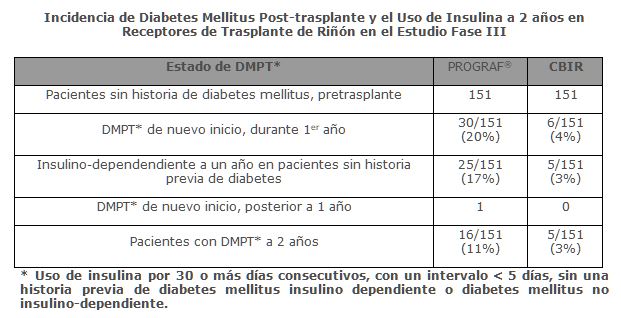
La diabetes mellitus insulino-dependiente post-trasplante se reportó en 18% y 11% de los pacientes con trasplante hepático tratados con PROGRAF® y fue reversible en 45% y 31% de estos pacientes a un año post-trasplante en los estudios aleatorios de EUA y Europa, respectivamente. La hiperglicemia se asoció al uso de PROGRAF® en 47% y 33% de los receptores de trasplante hepático en los estudios aleatorios de EUA y Europa, respectivamente, y puede requerir tratamiento.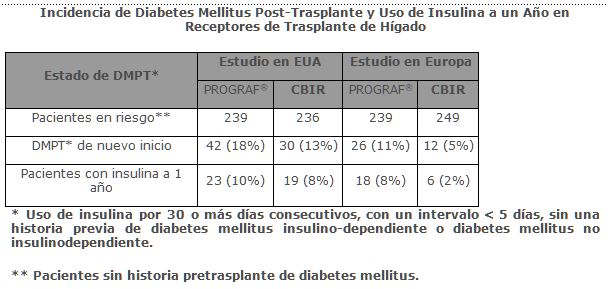
PROGRAF® puede causar nefrotoxicidad y neurotoxicidad, particularmente cuando se usa en dosis altas. La nefrotoxicidad se reportó en aproximadamente 52% de los pacientes con trasplante renal y en 40% y 36% de los pacientes con trasplante hepático que recibieron PROGRAF® en los estudios aleatorizados de EUA y Europa, respectivamente. La nefrotoxicidad se observa tempranamente después del trasplante y se caracteriza por aumento de la creatinina sérica y disminución de flujo urinario. Los pacientes con daño de la función renal deberán ser vigilados de cerca, ya que puede ser necesario reducir las dosis de PROGRAF®. En pacientes con elevaciones persistentes de creatinina sérica que no responden a los ajustes de dosis, se debe considerar el cambio a otra terapia inmunosupresora. Se debe tener cuidado al utilizar Tacrolimus con otros medicamentos nefrotóxicos. En especial para evitar exceso de nefrotoxicidad, PROGRAF® no deberá utilizarse simultáneamente con ciclosporina. PROGRAF® o ciclosporina deberán ser suspendidos por lo menos 24 horas antes de iniciar el otro. En presencia de concentraciones elevadas de PROGRAF® o ciclosporina la dosis con la nueva terapia, deberá de retardarse aún más. Se ha reportado hipercalemia leve a severa en 31% de los receptores de trasplante renal y en 45% y 13% en receptores de trasplante de hígado tratados con PROGRAF® en los estudios aleatorios en EUA y Europa, respectivamente, y pueden requerir tratamiento. Deben de vigilarse los niveles séricos de potasio y se debe evitar el uso de diuréticos ahorradores de potasio durante la terapia con PROGRAF®. En los dos estudios aleatorios se reportó en cerca de 55% de los receptores de trasplante hepático, neurotoxicidad incluyendo temblor, cefalea y otros cambios en la función motora, estado mental y la función sensitiva. El temblor se presentó más frecuentemente en los pacientes de trasplante renal tratados con PROGRAF® (54%) comparado con los pacientes tratados con ciclosporina. La incidencia de otros eventos neurológicos en los pacientes con trasplante renal fue similar en los dos grupos de tratamiento. El temblor y la cefalea han sido asociados con concentraciones altas de Tacrolimus en sangre completa y pueden responder a los ajustes de dosis. Han ocurrido convulsiones en pacientes adultos y pediátricos que recibían PROGRAF®. También se han asociado coma y delirio con altas concentraciones plasmáticas de tacrolimus. Al igual que en pacientes que reciben otros inmunosupresores, los pacientes que reciben PROGRAF® tienen un riesgo incrementado de desarrollar linfomas y otras malignidades, particularmente en la piel. El riesgo parece estar relacionado a la intensidad y duración de la inmunosupresión más que al uso de algún agente en específico. Un trastorno linfoproliferativo (TLP), relacionado con infección por virus de Epstein-Barr (EBV), se ha reportado en los receptores de trasplantes de órganos inmunosuprimidos. El mayor riesgo de TLP parece estar en niños pequeños que se encuentran en riesgo de infección con EBV primaria, mientras están inmunosuprimidos o quienes se han cambiado a PROGRAF® después de una terapia de inmunosupresión a largo plazo. Debido al peligro de sobre-supresión del sistema inmune, la cual puede aumentar la susceptibilidad a la infección, deberá utilizarse con precaución la terapia combinada de inmunosupresión. Algunos pacientes que han recibido PROGRAF® solución inyectable han experimentado reacciones anafilácticas. No obstante la causa exacta de estas reacciones no se conoce, otros medicamentos con derivados del aceite de ricino en la fórmula se han asociado con anafilaxia en un pequeño porcentaje de pacientes. Debido al riesgo potencial de anafilaxia, la inyección de PROGRAF® deberá reservarse para pacientes imposibilitados para tomar las cápsulas de PROGRAF®. Los pacientes que reciban PROGRAF® inyectado deberán estar bajo observación continua por lo menos, los primeros 30 minutos después de iniciar la infusión, y posteriormente a intervalos frecuentes. Si ocurrieran signos o síntomas de anafilaxia, se deberá detener la infusión. Deberá estar disponible una solución con epinefrina a la cabecera del paciente, así como una fuente de oxígeno. General: La hipertensión es un efecto adverso común de la terapia con PROGRAF®. La hipertensión leve o moderada, se reporta con más frecuencia que la hipertensión severa. Se puede requerir tratamiento antihipertensivo; el control de la presión arterial puede lograrse con cualquiera de los antihipertensivos comunes. Ya que Tacrolimus puede causar hipercalemia, deberán evitarse los diuréticos ahorradores de potasio. Mientras que los agentes bloqueadores de los canales del calcio pueden ser efectivos para tratar la hipertensión asociada a PROGRAF®, deberá tenerse cuidado ya que la interferencia con el metabolismo de Tacrolimus puede requerir una reducción de la dosis. Pacientes con daño renal y hepático: Para los pacientes con insuficiencia renal alguna evidencia sugiere utilizar dosis más bajas. El uso de PROGRAF® en receptores de trasplante de hígado que experimentan daño hepático post-trasplante puede estar asociado con el incremento del riesgo de desarrollar insuficiencia renal relacionada con altos niveles de Tacrolimus en sangre total. Los pacientes deben ser vigilados muy cercanamente y se debe considerar un ajuste de la dosis. Existe evidencia que sugiere que se deben utilizar dosis más bajas en estos pacientes. Pacientes con hipertrofia miocárdica: La hipertrofia miocárdica se ha reportado en asociación con la administración de PROGRAF® y generalmente se manifiesta por aumento concéntrico demostrado ecocardiográficamente de la pared posterior del ventrículo izquierdo y en el grosor del septum interventricular. Se ha observado hipertrofia en bebés, niños y adultos. Esta condición parece ser reversible en la mayoría de los casos, después de una reducción de la dosis o al suspender la terapia. En un grupo de 20 pacientes con ecocardiogramas pre- y post-tratamiento que mostraban evidencia de hipertrofia miocárdica, el promedio de las concentraciones de Tacrolimus en sangre total durante el periodo previo al diagnóstico de hipertrofia miocárdica se encontraban en un intervalo de 11 a 53 ng/ml en bebés (N = 10, edad 0.4 a 2 años), 4 a 46 ng/ml en niños (N = 7, edad 2 a 15 años) y 11 a 24 ng/ml en adultos (N = 3, edad 37 a 53 años). En pacientes que desarrollan insuficiencia renal o manifestaciones clínicas de disfunción ventricular mientras reciben terapia con PROGRAF®, debe considerarse una valoración ecocardiográfica. Si se diagnostica hipertrofia miocárdica debe considerarse una reducción de dosis o suspender PROGRAF®. Información para los pacientes: Los pacientes deberán estar informados de la necesidad de realizar pruebas de laboratorio apropiadas repetidamente, mientras están recibiendo PROGRAF®. Se les deberán dar instrucciones completas para la dosis, advertirlos de los riesgos potenciales durante el embarazo e informarlos del riesgo incrementado de neoplasia. Los pacientes deben estar informados de que los cambios en las dosis no deberán realizarse sin antes consultar a su médico. Los pacientes deberán estar informados que PROGRAF® puede causar diabetes mellitus y deberán ser orientados sobre la necesidad de acudir con su médico al aumentar la frecuencia en las micciones, aumento de sed o hambre. Así como con otros agentes inmunosupresores, debido al riesgo potencial de cambios malignos en la piel, se debe limitar la exposición a la luz del sol y a la radiación ultravioleta (UV) utilizando ropa protectora y bloqueador solar con un alto factor de protección.
Restricciones de uso durante el embarazo y la lactancia: En los estudios de reproducción en ratas y conejos, se observaron efectos adversos en el feto, principalmente a niveles en la dosis que eran tóxicos para las madres. Dosis orales de Tacrolimus de 0.32 y 1.0 mg/kg durante la organogénesis en conejos se asociaron con toxicidad materna, así como con un incremento en la incidencia de abortos, estas dosis son equivalentes a 0.5-1X y 1.6-3.3X el intervalo de dosis clínicamente recomendado (0.1 a 0.2 mg/kg) basado en las correcciones del área de la superficie del cuerpo. Solamente a las dosis más altas se ha observado un incremento en la incidencia de malformaciones y alteraciones en el desarrollo. Dosis orales de 3.2 mg/kg de Tacrolimus durante la organogénesis en ratas, se han asociado con toxicidad materna y causado un incremento de la resorción tardía, disminución en el número de nacimientos vivos, así como en el peso y viabilidad de las crías. La administración oral de Tacrolimus, a 1.0 y 3.2 mg/kg (equivalente a 0.7-1.4X y 2.3-4.6X de la dosis clínicamente recomendada con base en una corrección del área de la superficie del cuerpo), a ratas preñadas después de la organogénesis y durante la lactancia se asoció con crías de bajo peso. No se demostró disminución de fertilidad en hombres o mujeres. No existen estudios adecuados y bien controlados en mujeres embarazadas. Tacrolimus cruza la placenta. El uso de tacrolimus durante el embarazo ha sido asociado con hipercalemia neonatal y disfunción renal. PROGRAF® podrá ser usado durante el embarazo sólo si el beneficio potencial a la madre justifica el riesgo potencial del feto. Lactancia: Tacrolimus es excretado por la leche humana, por lo tanto, se debe evitar durante la lactancia.Pacientes pediátricos: La experiencia con PROGRAF® en pacientes pediátricos con trasplante de riñón es limitada. Se han realizado trasplantes de hígado exitosamente en pacientes pediátricos (mayores de 16 años) utilizando PROGRAF®. Dos estudios aleatorios activamente controlados de PROGRAF® en trasplante primario de hígado incluyeron 56 pacientes pediátricos. 31 pacientes se asignaron aleatoriamente a la terapia basada en PROGRAF® y 25 a CBIR. Además, un mínimo de 122 pacientes pediátricos fueron estudiados en un análisis no controlado de Tacrolimus en trasplante de hígado de donador vivo relacionado. Los pacientes pediátricos generalmente requirieron dosis más altas de PROGRAF® para mantener las concentraciones de Tacrolimus en sangre total similares a las de los pacientes adultos. No se use durante el embarazo y la lactancia.
Reacciones secundarias y adversas: Trasplante de hígado: Las principales reacciones adversas de PROGRAF® son temblor, dolor de cabeza, diarrea, hipertensión, náusea y disfunción renal. Estos ocurren con la administración oral o IV de PROGRAF® y pueden responder a la reducción de la dosis. La diarrea fue asociada algunas veces con otras molestias gastrointestinales como náusea y vómito. Hipercalemia e hipomagnesemia se han presentado en pacientes que reciben terapia con PROGRAF®. Se ha observado hiperglucemia en muchos pacientes, algunos pueden requerir terapia con insulina. La incidencia de los eventos adversos fue determinada en dos estudios de trasplante de hígado, comparativos, aleatorios entre 514 pacientes que recibieron Tacrolimus y esteroides y 515 pacientes que recibieron un régimen basado en ciclosporina (CBIR). La proporción de los pacientes que reportaron más de un evento adverso fue de 99.8% en el grupo de Tacrolimus y 99.6% en el grupo CBIR. Se deben tomar precauciones al comparar la incidencia de efectos adversos en el estudio de EUA contra el estudio de Europa. La información post-trasplante de 12 meses del estudio de EUA y del estudio de Europa se presentan abajo. Los dos estudios incluyen también diferentes poblaciones de pacientes y pacientes que fueron tratados con regímenes de inmunosupresores de diferentes intensidades. Los eventos adversos ≥ 15% reportados en pacientes con Tacrolimus (resultados de estudio combinados) se presentan abajo para los dos estudios controlados en trasplante de hígado.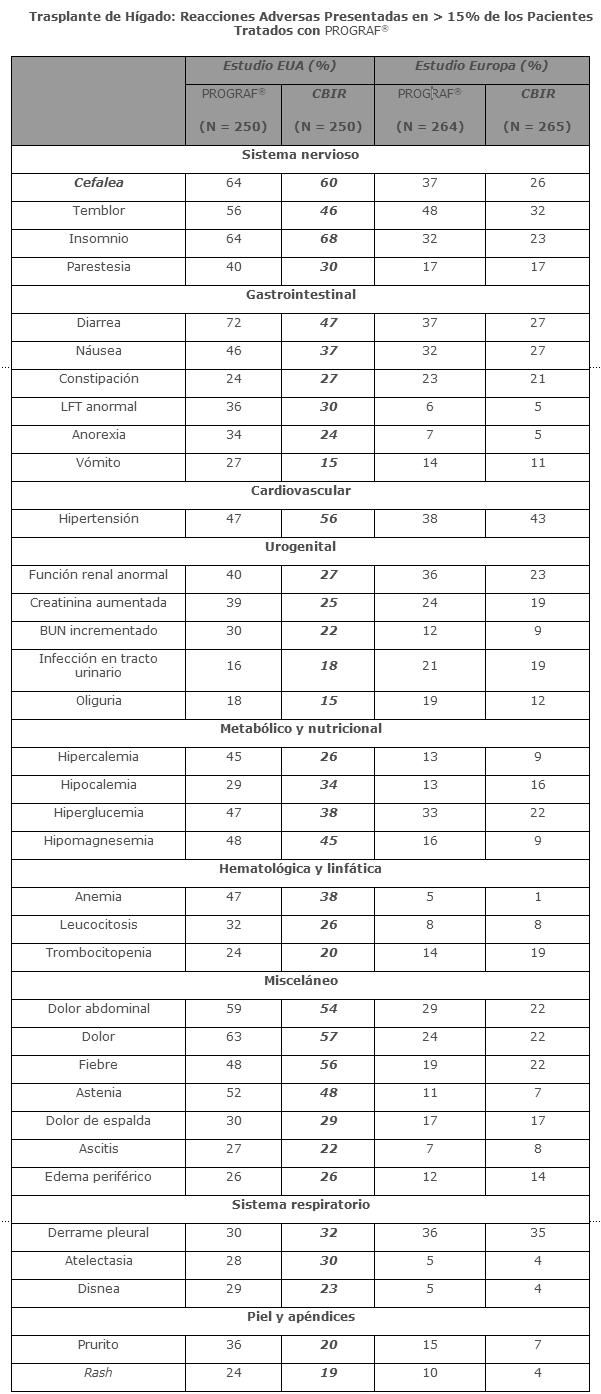
Las reacciones adversas observadas menos frecuentemente en pacientes con trasplante de hígado y riñón se describen más adelante. Trasplante de riñón: Las reacciones adversas más comúnmente reportadas fueron infección, temblor, hipertensión, disminución de la función renal, constipación, diarrea, dolor de cabeza, dolor abdominal e insomnio. Las reacciones adversas que ocurrieron en ≥ 15% de pacientes con trasplante de riñón tratados con PROGRAF® se presentan abajo.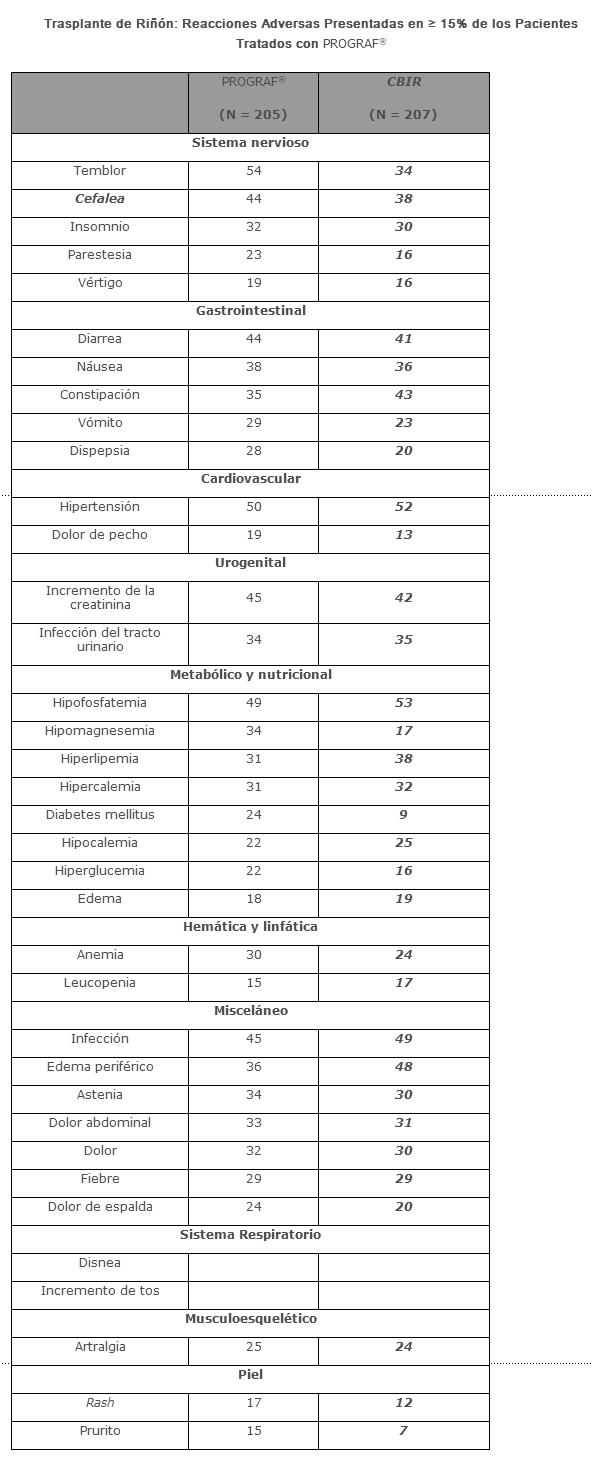
Las reacciones adversas observadas menos frecuentemente en pacientes con tra splante de hígado y riñón se describen más adelante. Reacciones adversas menos frecuentemente reportadas: Los siguientes eventos adversos fueron reportados por receptores de trasplante de hígado o riñón que fueron tratados con Tacrolimus en estudios clínicos. Sistema Nervioso: Sueños anormales, agitación, amnesia, ansiedad, confusión, convulsión, llanto, depresión, vértigo, manía, labilidad emocional, encefalopatía, apoplejía hemorrágica, alucinaciones, hipertonía, incoordinación, monoparesia, mioclonías, compresión del nervio, nerviosismo, neuropatía, parálisis flácida, habilidades psicomotoras disminuidas, psicosis, cuadriparesia, somnolencia, pensamientos anormales, escritura disminuida. Sentidos especiales: Visión anormal, ambliopía, dolor de oído, otitis media, tinnitus. Sistema gastrointestinal: Anorexia, colangitis, ictericia colestática, duodenitis, dispepsia, disfagia, esofagitis, flatulencia, gastritis, gastroesofagitis, hemorragia gastrointestinal, incremento de TGG, perforación GI, hepatitis, hepatitis granulomatosa, íleo, incremento del apetito, ictericia, daño hepático, pruebas de función hepáticas anormales, esofagitis ulcerativa, moniliasis oral, pseudoquiste pancreático, trastorno rectal, estomatitis. Cardiovascular: Angina de pecho, fibrilación cardiaca, falla cardiopulmonar, dolor torácico, tromboflebitis profunda, ECG anormal, ecocardiograma anormal, complejo QRS anormal en el electrocardiograma, segmento ST anormal en el electrocardiograma, disminución del ritmo cardiaco hemorragia, hipotensión, hipotensión postural, trastorno vascular periférico, flebitis, taquicardia, trombosis, vasodilatación. Urogenital: Albuminuria, espasmo de la vejiga, cistitis, disuria, hematuria, hidronefrosis, falla renal, necrosis tubular renal, nocturia, oliguria, piuria, nefropatía tóxica, incontinencia urgente, frecuencia urinaria, incontinencia urinaria, retención urinaria y vaginitis. Metabólico/nutricional: Acidosis, fosfatasa alcalina incrementada, alcalosis, TGO incrementada, TGP incrementada, disminución del bicarbonato, bilirrubinemia, BUN incrementado, deshidratación, GGT incrementada, curación anormal, hipercalcemia, hipercolesterolemia, hiperlipidemia, hiperfosfatemia, hiperuricemia, hipervolemia, hipocalcemia, hipoglucemia, hiponatremia, hipofosfatemia, hipoproteinemia, incremento de la deshidrogenasa láctica, ganancia de peso. Endocrino: Síndrome de Cushing, diabetes mellitus. Hemático/Linfático: Trastornos de la coagulación, equimosis, incremento del hematócrito, hemoglobina anormal, anemia hipocrómica, leucocitosis, leucopenia, policitemia, disminución de la protrombina, disminución del hierro sérico, trombocitopenia. Misceláneo: Distensión abdominal, abscesos, daño accidental, reacción alérgica, celulitis, calosfríos, síndrome gripal, recaídas, sensación de anormalidad, edema generalizado, hernia, disminución de la movilidad, peritonitis, reacción de fotosensibilidad, sepsis, intolerancia a la temperatura, úlcera. Musculoesquelético: Artralgia, calambres, espasmo generalizado, trastorno articular, calambres en las piernas, mialgia, miastenia, osteoporosis. Respiratorio: Asma, bronquitis, incremento en la tos, enfisema, hipo, trastorno pulmonar, neumotórax, edema pulmonar, faringitis, neumonía, trastorno respiratorio, rinitis, sinusitis, alteración de la voz. Piel: Acné, alopecia, dermatitis exfoliativa, dermatitis micótica, herpes simple, hirsutismo, decoloración de la piel, trastorno de la piel, úlcera cutánea, sudoración. Experiencia Post-comercialización: Eventos adversos post-comercialización: Los siguientes eventos adversos han sido reportados en la venta de PROGRAF® a nivel mundial. Debido a que estos efectos se reportan de manera voluntaria de una población de tamaño desconocido, pueden estar asociados con otras enfermedades, terapias de fármacos múltiples y procedimientos quirúrgicos no siempre es posible estimar su frecuencia o establecer una causa relacionada a la exposición al fármaco. La decisión para incluir estos efectos está basada típicamente en uno o más de los siguientes factores: (1) gravedad del evento, (2) frecuencia del reporte, (3) conexión causal con el fármaco de validez. Raramente ha habido reportes espontáneos de hipertrofia miocárdica asociada con manifestaciones clínicas de disfunción ventricular en pacientes que están recibiendo terapia con PROGRAF®. Otros efectos: Cardiovascular: Fibrilación auricular, flutter, arritmia cardiaca, paro cardiaco, electrocardiograma con onda T anormal, ruborizaciones, infarto del miocardio, isquemia miocárdica, derrame pericárdico, prolongación de la QT, Torsade de pointes, trombosis venosa profunda en extremidades, extrasístoles ventriculares y fibrilación ventricular. Gastrointestinales: Estenosis del conducto biliar, colitis, enterocolitis, gastroenteritis, enfermedad de reflujo gastroesofágico, citolisis hepática, necrosis hepática, hepatotoxicidad, vaciamiento gástrico disfuncional, hígado graso, úlceras bucales, pancreatitis hemorrágica, pancreatitis necrotizante, úlcera estomacal, enfermedad del hígado venooclusiva. Hemática/linfática: Coagulación intravascular diseminada, neutropenia, pancitopenia, púrpura trombocitopénica, púrpura trombocitopénica trombótica. Metabólico/nutricional: Glucosuria, incremento de la amilasa incluyendo pancreatitis, disminución de peso. Misceláneo: Escalofríos, ansiedad, bochornos, falla multiorgánica, disfunción primaria del trasplante. Sistema Nervioso: Síndrome del túnel carpiano, infarto cerebral, hemiparesia, leucoencefalopatía, desorden mental, mutismo, cuadriplejía, desorden del habla, síncope. Respiratorio: Síndrome de dolor respiratorio agudo, infiltración pulmonar, dolor respiratorio, falla respiratoria. Piel: Síndrome de Stevens-Johnson, necrólisis epidérmica tóxica. Sentidos especiales: Ceguera, ceguera cortical, disminución de la audición incluyendo sordera, fotofobia. Urogenital: Falla renal aguda, cistitis hemorrágica, síndrome urémico-hemolítico, desorden en la micción.
Interacciones medicamentosas y de otro género: Interacciones con otros fármacos: Debido al potencial aditivo o sinérgico en el daño de la función renal, se debe tener cuidado cuando se administre PROGRAF® con otros fármacos que puedan estar asociados con disfunción renal. Esto incluye, pero no limita a, aminoglucósidos, anfotericina B y cisplatino. La experiencia clínica inicial en la co-administración de PROGRAF® y ciclosporina provocó un efecto aditivo/sinérgico de nefrotoxicidad. En pacientes que cambian de ciclosporina a terapia con PROGRAF® deberán recibir la primera dosis de PROGRAF® después de 24 horas de la última dosis de ciclosporina. La dosificación debe retardarse en presencia de niveles elevados de ciclosporina. Fármacos que pueden afectar las concentraciones de Tacrolimus: Debido a que tacrolimus es metabolizado principalmente por el sistema de enzimas CYP3A, las sustancias que se sabe inhiben estas enzimas pueden disminuir el metabolismo o incrementar la biodisponibilidad de tacrolimus como lo demuestra la disminución de las concentraciones en sangre total o plasma. La vigilancia de las concentraciones sanguíneas y los ajustes de dosis necesarios, son esenciales cuando estos medicamentos se utilizan simultáneamente.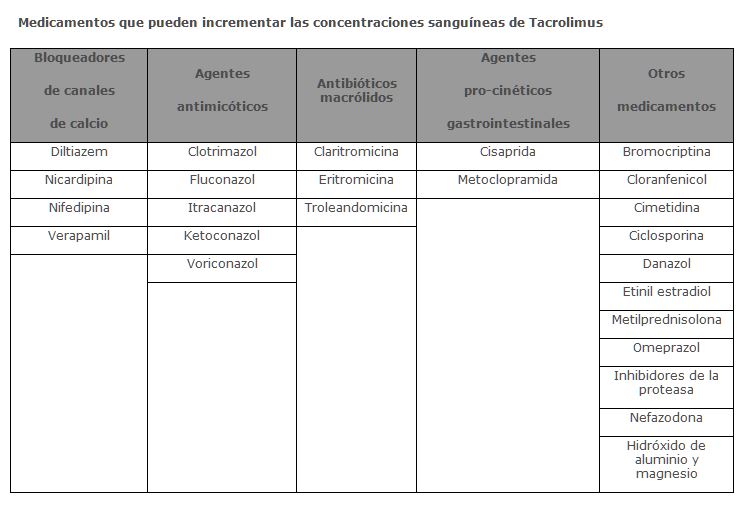
En un estudio con 6 voluntarios normales se observó un incremento significativo en la biodisponibilidad oral de Tacrolimus (14.5 ± 5% contra 30 ± 8%) al administrar simultáneamente ketoconazol (200 mg). La depuración oral de Tacrolimus durante la administración de ketoconazol disminuyó significativamente comparado con Tacrolimus solo (0.430 ± 0.129 L/hr/kg contra 0.148 ± 0.043 L/hr/kg); sin embargo, la depuración IV de Tacrolimus no cambió significativamente al ser administrado al mismo tiempo que ketoconazol, aunque estos resultados variaron ampliamente entre pacientes.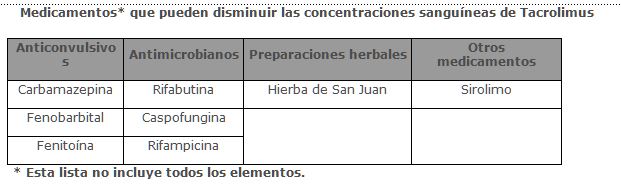
La hierba de San Juan (Hypericum perforatum) induce a la CYP3A4 y glucoproteína-P. Debido a que tacrolimus es un sustrato para CYP3A4 existe la probabilidad de que el uso de la hierba de San Juan en pacientes que reciben PROGRAF® pueda producir la reducción de los niveles de tacrolimus. En un estudio cruzado de dosis única en voluntarios sanos, al administrar al mismo tiempo Tacrolimus e hidróxido de magnesio y aluminio se produjo un incremento del 21% en el promedio AUC de Tacrolimus y una disminución del 10% en el promedio de Cmax de Tacrolimus respecto a la administración de Tacrolimus solo. En un estudio con 6 voluntarios normales se observó una disminución significativa en la biodisponibilidad oral (14 ± 6% contra 7 ± 3%) al administrar simultáneamente rifampina (600 mg). Además hubo un incremento significativo en la depuración de Tacrolimus (0.036 ± 0.008 L/hr/kg contra 0.053 ± 0.010 L/hr/kg) durante la administración simultánea de rifampina. No se han realizado estudios de interacción con fármacos usados en el tratamiento de VIH. Sin embargo, se debe tener cuidado cuando se requieran administrar simultáneamente fármacos que son nefrotóxicos (por ejemplo, ganciclovir) o que pueden ser metabolizados por CYP3A, (por ejemplo, nelfinavir, ritonavir) al ser administrados simultáneamente con Tacrolimus. Basado en un estudio clínico de 5 receptores de trasplante de hígado, la administración simultánea de Tacrolimus con nelfinavir incrementó significativamente las concentraciones de Tacrolimus en sangre, y como resultado, fue necesario la reducción de la dosis de Tacrolimus en un promedio de 16 veces para mantener las concentraciones en sangre en 9.7 ng/ml. Por lo