PROLIA®
AMGEN
Denominación genérica: Denosumab.
Forma farmacéutica y formulación: Solución. Inyección subcutánea. Cada jeringa prellenada contiene: Denosumab 60 mg. Vehiculo c.s 1.0 mL.
Indicaciones terapéuticas: Osteoporosis posmenopáusica: PROLIA está indicado en el tratamiento de la osteoporosis en mujeres posmenopáusicas. En las mujeres posmenopáusicas con osteoporosis, PROLIA incrementa la densidad mineral ósea (DMO) y reduce la incidencia de fracturas de cadera, vertebrales y no vertebrales. Pérdida ósea en pacientes que se someten a ablación hormonal por cáncer: PROLIA está indicado para el tratamiento de la pérdida ósea en pacientes que se someten a ablación hormonal para cáncer de próstata o cáncer de mama. En los pacientes con cáncer de próstata, PROLIA reduce la incidencia de fracturas vertebrales.
Farmacocinética y farmacodinamia: Propiedades farmacocinéticas: Después de la administración subcutánea, PROLIA mostró una farmacocinética no lineal con una dosis sobre un amplio intervalo de dosis e incrementos proporcionales para la dosis expuesta de 60 mg (o 1 mg/kg) y superiores. Absorción: Después de una dosis subcutánea de 60 mg de denosumab, la biodisponibilidad fue de 61% y las concentraciones máximas de denosumab en el suero (Cmax) de 6 mg/mL (rango de 1 a 17 mg /mL) ocurrieron en 10 días (rango de 2 a 28 días). Después de que los niveles séricos de Cmax declinaran con una vida media de 26 días (rango de 6 a 52 días) durante un período de 3 meses (rango de 1.5 a 4.5 meses). Cincuenta y tres por ciento de los pacientes no presentaron cantidades medibles detectables de denosumab a los 6 meses posteriores a la administración. Distribución: No se observó una acumulación o cambio en el tiempo, en la farmacocinética de PROLIA con dosis múltiples subcutáneas de 60 mg una vez cada seis meses. Metabolismo: El denosumab está compuesto únicamente de aminoácidos y carbohidratos como la inmunoglobulina nativa. Con base en los datos no clínicos, el metabolismo del denosumab está previsto para seguir las rutas de eliminación de la inmunoglobulina, teniendo como resultado la degradación en péptidos pequeños y aminoácidos individuales. Eliminación: El denosumab está compuesto únicamente de aminoácidos y carbohidratos como la inmunoglobulina nativa y no está previsto que se elimine a través de los mecanismos metabólicos hepáticos (por ejemplo, enzimas del citocromo p450 (CYP)). Con base en los datos no clínicos, su eliminación está prevista que siga las rutas de eliminación de la inmunoglobulina, teniendo como resultado la degradación en péptidos pequeños y aminoácidos individuales. En un estudio de 17 mujeres postmenopáusicas con osteoporosis, se administró midazolam (2 mg oral) dos semanas después de una sola dosis de denosumab (60 mg SC), que corresponde al tiempo de los efectos farmacodinámicos máximos de denosumab. Denosumab no afectó la farmacocinética de midazolam, que se metaboliza por el citocromo P450 3A4 (CYP3A4). Esto indica que denosumab no debe alterar la PK de los fármacos metabolizados por el CYP3A4. Poblaciones de pacientes especiales:Ancianos (mayores a 65 años): No se encontró que la edad fuera un factor considerable en la farmacocinética del denosumab en un análisis farmacocinético de la población de pacientes en un rango de edad de 28 a 87 años. Niños y adolescentes (hasta 18 años): No existen datos farmacocinéticos disponibles en pacientes pediátricos. Raza: La farmacocinética del denosumab no fue afectada por la raza en mujeres posmenopáusicas o en pacientes con cáncer de mama sometidos a ablación hormonal. Insuficiencia renal: En un estudio de 55 pacientes con varios grados de función renal, incluyendo pacientes en diálisis, el grado de insuficiencia renal no tuvo un efecto en la farmacocinética y farmacodinámica del denosumab; por lo tanto, no es necesario el ajuste de la dosis para la insuficiencia renal. Insuficiencia hepática: No se han llevado a cabo estudios clínicos para evaluar el efecto de la insuficiencia hepática en la farmacocinética del denosumab. Farmacodinamia: Mecanismo de acción: El denosumab es un anticuerpo monoclonal humano (IgG2) que se dirije y se une con una alta afinidad y especificidad al RANKL (Ligando del Receptor Activador del Factor Nuclear k B) previniendo que el RANKL active su único receptor, RANK, sobre la superficie de osteoclastos y sus precursores, independientemente de la superficie ósea. La prevención de la interacción RANKL/RANK inhibe la formación, funciones y sobrevivencia de los osteoclastos. Por lo tanto, el denosumab reduce la resorción ósea e incrementa la masa y resistencia óseas tanto en el hueso cortical y trabecular. Efectos farmacodinámicos: En estudios clínicos, el tratamiento con 60 mg de denosumab tuvo como resultado la reducción rápida en suero del marcador de resorción ósea tipo 1 C-telopéptidos (CTX) en las 6 horas posteriores a la administración subcutánea (en aproximadamente 70%) con reducciones de aproximadamente 85% las cuales ocurrieron en 3 días. Las reducciones de CTX se mantuvieron durante el intervalo de dosis de 6 meses. Al final de cada intervalo de dosis, las reducciones de CTX se atenuaron parcialmente a partir de la reducción máxima ≥ 87% hasta aproximadamente ≥45% (en un rango de 45 a 80%), reflejando así la reversibilidad de los efectos del denosumab en la remodelación ósea una vez que disminuyen los niveles séricos. Estos efectos se sostuvieron con el tratamiento continuo. En concordancia con la relación fisiológica de la formación y resorción ósea en la remodelación del esqueleto, las reducciones en los marcadores de formación ósea (por ejemplo se observaron la fosfatasa alcalina específica del hueso [BSAP, por sus siglas en inglés] y el propéptido N-terminal en suero de colágeno tipo 1 [P1NP]) después de un mes de la primera dosis de denosumab. Los marcadores de recambio óseo (marcadores de resorción y formación ósea) alcanzaron generalmente niveles de pretratamiento previo en un máximo de 9 meses después de la última dosis subcutánea de 60 mg. Al reiniciar, el grado de inhibición de CTX por denosumab fue similar al observado en pacientes al iniciar el tratamiento con denosumab. En un estudio clínico de mujeres posmenopáusicas con masa ósea baja (N = 504) las cuales recibieron un tratamiento previo con alendronato durante una duración promedio de 3 años, aquellas que cambiaron a denosumab experimentaron reducciones adicionales en CTX en suero, en comparación con las mujeres que permanecieron con alendronato. En este estudio los cambios en el calcio sérico fueron similares entre los dos grupos. Immunogenicidad: El denosumab es un anticuerpo monoclonal humano; como con todas las proteínas terapéuticas, existe un potencial téorico de inmunogenicidad. Más de 13000 pacientes fueron evaluados para anticuerpos vinculantes mediante el uso de un inmunoensayo de electroquimioluminiscencia sensible. Menos del 1% de los pacientes con tratamiento de denosumab hasta por 5 años, resultaron positivos (incluyendo anticuerpos previamente existentes, transitorios y en desarrollo). Los pacientes que resultaron positivos para anticuerpos vinculantes se evaluaron adicionalmente para anticuerpos neutralizantes mediante un ensayo biológico in vitro basado en células quimioluminiscentes y ninguno de ellos resultó positivo. Ninguna evidencia de alteración en el perfil farmacocinético, perfil de toxicidad o respuesta clínica se asoció con el desarrollo de anticuerpos vinculantes. Estudios clínicos: Tratamiento de la osteoporosis posmenopáusica: Se demostraron la eficacia y seguridad del denosumab en el tratamiento de la osteoporosis posmenopáusica en FREEDOM, un estudio de 3 años, aleatorizado, doble ciego, controlado con placebo, multinacional que demostró que el denosumab fue efectivo en comparación con el placebo al reducir las nuevas fracturas vertebrales, no vertebrales y de cadera en mujeres posmenopáusicas con osteoporosis. 7,808 mujeres con edades de 60 a 91 años se enrolaron, de las cuales el 23.6% tenía fracturas vertebrales prevalentes. Las mujeres fueron aleatorizadas para recibir inyecciones subcutáneas ya sea de placebo (n = 3,906) o denosumab 60 mg (n = 3,902) una vez cada 6 meses. Las mujeres recibieron complementos diarios de calcio (al menos 1,000 mg) y vitamina D (al menos 400 UI). La variable primaria de eficacia fue la incidencia de nuevas fracturas vertebrales. Las variables secundarias de eficacia incluyeron la incidencia de fracturas no vertebrales y fracturas de cadera, evaluadas después de 3 años. El denosumab redujo considerablemente el riesgo de nuevas fracturas vertebrales, no vertebrales y de cadera en comparación con el placebo. Los 3 criterios de valoración de eficacia de fracturas obtuvieron el nivel de significancia estadística con base en el esquema de pruebas secuenciales preespecificadas. Efecto en las fracturas vertebrales: El denosumab redujo considerablemente el riesgo de nuevas fracturas vertebrales (criterio de valoración primario) en un 68% (tasa de riesgo: 0.32; p < 0.0001) durante 3 años. Las tasas de fractura después de 3 años para las nuevas fracturas vertebrales fueron de 7.2% en el grupo de placebo y 2.3% en el grupo de PROLIA (reducción de riesgo absoluto no ajustado de 4.8%). Las reducciones también se observaron desde el primer año (61% de reducción de riesgo relativo; 1.4% de reducción de riesgo absoluto no ajustado) y el segundo año de tratamiento (71% de reducción de riesgo relativo; 3.5% de reducción de riesgo absoluto no ajustado) (todas las p < 0.0001). El denosumab también redujo el riesgo de otras categorías de fracturas preespecificadas, incluyendo fracturas vertebrales nuevas o en deterioro (67% de reducción de riesgo relativo, 4.8% de reducción de riesgo absoluto no ajustado), nuevas fracturas vertebrales múltiples (61% de reducción de riesgo relativo, 1.0% de reducción de riesgo absoluto no ajustado), fracturas vertebrales clínicas (69% de reducción de riesgo relativo, 1.8% de reducción de riesgo absoluto no ajustada) durante 3 años. Las reducciones en el riesgo de nuevas fracturas vertebrales mediante denosumab durante 3 años fueron consistentes y considerables independientemente del riesgo de fractura basal a 10 años evaluado por por el FRAX® (herramienta para la evaluación del riesgo de fractura de la OMS) independientemente de si las mujeres tuvieron fracturas vertebrales prevalentes o con historial de fracturas no vertebrales, sin importar la edad, DMO, nivel de recambio óseo y uso previo de un medicamento para la osteoporosis en línea basal. En mujeres posmenopáusicas con osteoporosis en edades mayores de 75 años, el denosumab redujo la incidencia de nuevas fracturas vertebrales (64%) y no vertebrales (16%). Efecto en todas las fracturas clínicas: El denosumab redujo considerablemente el riesgo de fracturas no vertebrales (criterio de valoración secundario) en 20% (tasa de riesgo: 0.80; p = 0.0106) durante 3 años. Las tasas de fracturas no vertebrales fueron de 8.0% en el grupo de placebo y de 6.5% en el grupo de denosumab (reducción de riesgo absoluto no ajustado de 1.5%). El denosumab también redujo el riesgo de fracturas clínicas (30% de reducción de riesgo relativo, 2.9% de reducción de riesgo absoluto no ajustado), fracturas no vertebrales graves (20% de reducción de riesgo relativo, 1.2% de reducción de riesgo absoluto no ajustado) y las fracturas osteoporóticas graves (35% de reducción de riesgo relativo, 2.7% de reducción de riesgo absoluto no ajustado) durante 3 años. En mujeres con un valor T de DMO de cuello femoral de línea basal menor o igual a -2.5, el denosumab redujo la incidencia de fracturas no vertebrales (35% de reducción de riesgo relativo, 4.1% de reducción de riesgo absoluto no ajustado, p < 0.001) durante 3 años. Las reducciones en las fracturas no vertebrales se observaron independientemente de la probabilidad de línea basal a 10 años de una fractura osteoporótica grave como se evaluó mediante FRAX®. Efecto en las fracturas de cadera: Denosumab redujo considerablemente el riesgo de fracturas de cadera (criterio de valoración secundario) en 40% (tasa de riesgo: 0.60; p = 0.0362) durante 3 años. Las tasas de fractura de cadera a tres años fueron de 1.2% en el grupo de placebo y 0.7% en el grupo de denosumab (reducción de riesgo absoluto no ajustado de 0.5%). Las reducciones en el riesgo de fracturas de cadera durante 3 años fueron consistentes y considerablemente independientes a la probabilidad en la línea basal a 10 años de una fractura de cadera como se evaluó mediante FRAX®. En mujeres con alto riesgo de fractura como se definió anteriormente mediante la edad en la línea basal, la DMO y las fracturas vertebrales prevalentes, se observó un 48% de reducción del riesgo relativo con denosumab (1.1% de reducción de riesgo absoluto no ajustada). En un análisis post-hoc en mujeres posmenopáusicas con osteoporosis en edades mayores de 75 años, el denosumab redujo la incidencia de fracturas de cadera (62%). Efecto en la densidad mineral ósea (DMO): El denosumab incrementó considerablemente la DMO en todos los sitios clínicos analizados, comparado con placebo después de 1, 2 y 3 años. El denosumab incrementó la DMO en 9.2% en la columna lumbar, 6.0% en la cadera total, 4.8% en el cuello femoral, 7.9% en el trocanter de la cadera, 3.5% en el 1/3 distal del radio y 4.1% en el cuerpo total durante 3 años. Los incrementos en la DMO en la columna lumbar, cadera total y trocanter de cadera se observaron tan rápido como 1 mes posterior a la dosis inicial. El denosumab incrementó la DMO de la columna lumbar a partir de la línea basal en 96% de las mujeres posmenopáusicas después de 3 años. Se observaron efectos consistentes en la DMO de la columna lumbar independientemente de la edad, raza, peso, IMC, DMO y nivel de remodelado óseo en la línea basal. Histología ósea: Las evaluaciones de histología mostraron huesos de arquitectura y calidad normales, así como la reducción prevista en remodelado óseo comparado con placebo. No hubo evidencia de defectos de mineralización, tejido fibroso o fibrosis medular. Datos clínicos comparativos vs alendronato en el tratamiento de mujeres posmenopáusicas con masa ósea baja: En dos estudios aleatorizados, doble ciego, controlados activamente, uno en mujeres sin tratamiento previo y el otro en mujeres con un tratamiento previo con alendronato, el denosumab mostró incrementos considerablemente mayores en la DMO y reducciones en los marcadores de recambio óseo (por ejemplo, CTX en suero), en comparación con el alendronato. Se observaron incrementos consistentemente mayores en la DMO en la columna lumbar, cadera total, cuello femoral, trocanter de cadera y 1/3 distal del radio en mujeres que recibieron tratamiento con denosumab, en comparación con aquellas que continuaron recibiendo terapia con alendronato (todos p < 0.05). Eficacia clínica en el tratamiento de la pérdida ósea asociada con la ablación hormonal: Tratamiento de la pérdida ósea asociada con la deprivación androgénica: La eficacia y seguridad del denosumab en el tratamiento de la pérdida ósea asociada con la deprivación androgénica se evaluaron en un estudio de 3 años, aleatorizado, doble ciego, contolado con placebo, multinacional de 1,468 hombres con cáncer de próstata no metastático con edades de 48 a 97 años. Los hombres menores a 70 años de edad también presentaron ya sea un valor T de DMO en la columna lumbar, cadera total o de cuello femoral menor a -1.0 o un historial de alguna fractura osteoporótica. Los sujetos recibieron ya sea inyecciones subcutáneas de denosumab 60 mg (n = 734) o placebo (n = 734) una vez cada 6 meses. Los hombres recibieron complementos diarios de calcio (al menos 1,000 mg) y vitamina D (al menos 400 UI). Se observaron incrementos considerables en la DMO de la columna lumbar, cadera total, cuello femoral y el trocanter de la cadera tan rápido como 1 mes después de la dosis inicial. El denosumab incrementó la DMO de la columna lumbar en 7.9%, DMO de cadera total en 5.7%, DMO de cuello femoral en 4.9%, DMO de trocanter de cadera en 6.9%, DMO de 1/3 distal de radio en 6.9%, y DMO del cuerpo total en 4.7% durante 3 años, en comparación con placebo (P < 0.0001). Se observaron efectos consistentes en la DMO de la columna lumbar independientemente de la edad, raza, región geográfica, peso/IMC, DMO, nivel de recambio óseo; duración de la deprivación androgénica y presencia de fractura vertebral en la línea basal. El denosumab redujo considerablemente el riesgo de nuevas fracturas vertebrales en 62% (tasa de riesgo: 0.38; p < 0.0063) durante 3 años. También se observaron reducciones durante un año (85% reducción de riesgo relativo; 1.6% reducción de riesgo absoluto) y después de 2 años (69% de reducción de riesgo relativo; 2.2% reducción de riesgo absoluto) (todos p < 0.01). El denosumab también redujo la incidencia de los sujetos para más de una fractura osteoporótica en cualquier sitio en 72% comparado con placebo durante 3 años (placebo 2.5% vs denosumab 0.7%, p = 0.0063). Tratamiento de la pérdida ósea en mujeres que se sometieron a una terapia de inhibidor de aromatasa para cáncer de mama: Se evaluaron la eficacia y seguridad del denosumab en el tratamiento de la pérdida ósea asociada con la terapia adyuvante de inhibidor de aromatasa en un estudio de 2 años, aleatorizado, doble ciego, controlado por placebo, multinacional de 252 mujeres con cáncer de mama no metastático con edades de 35 a 84 años. Las mujeres presentaron valores T de DMO en la línea basal entre -1.0 a -2.5 en la columna lumbar, cadera total o cuello femoral. Las mujeres se aleatorizaron para recibir inyecciones subcutáneas ya sea de denosumab de 60 mg (n = 127) ó placebo (n = 125) una vez cada 6 meses. Las mujeres recibieron un complemento diario de calcio (al menos 1,000 mg) y vitamina D (al menos 400 UI). La variable de eficacia primaria fue el cambio porcentual en la DMO de la columna lumbar. El denosumab incrementó considerablemente la DMO en todos los sitios clínicos analizados, comparado con placebo a 2 años: 7.6% en la columna lumbar, 4.7% en la cadera total, 3.6% en el cuello femoral, 5.9% en el trocanter de cadera, 6.1% en el 1/3 distal del radio y 4.2% en el cuerpo total. Se observaron incrementos considerables en la DMO de la columna lumbar tan rápido com al mes de la dosis inicial. Se observaron efectos consistentes en la DMO independientemente de la edad, duración de la terapia con inhibidor de aromatasa, peso /IMC, quimioterapia previa, uso previo de moduladores selectivos del receptor de estrógeno (SERM por sus siglas en inglés), y tiempo desde la menopausia.
Contraindicaciones: Hipersensibilidad a denosumab o a los componentes de PROLIA. Hipocalcemia.
Precauciones generales: El consumo adecuado de calcio y vitamina D es importante en todos los pacientes que reciben PROLIA. La hipocalcemia debe corregirse por medio de un consumo adecuado de calcio y vitamina D antes del inicio de la terapia. Se recomienda el monitoreo clínico de los niveles de calcio para los pacientes con una predisposición a la hipocalcemia (consulte reacciones adversas). Los pacientes que reciben PROLIA pueden desarrollar infecciones en la piel (predominantemente celulitis) que pueden llevarlos a la hospitalización. Estos eventos se notificaron con mayor frecuencia en los grupos de denosumab (0.4%) vs el placebo (0.1%) (consulte Reacciones secundarias y adversas). La incidencia general de infecciones en la piel fue similar entre los grupos de placebo y denosumab. Se debe asesorar a los pacientes para buscar atención médica inmediata en caso de desarrollar señales o síntomas de celulitis. Los casos de osteonecrosis mandibular (OM) se notificaron predominantemente en pacientes con cáncer avanzado los cuales recibían 120 mg cada 4 semanas. La OM se notificó con poca frecuencia en pacientes con osteoporosis que recibían 60 mg cada 6 meses (consulte Reacciones secundarias y adversas). Fracturas femorales atípicas han sido reportados en pacientes que reciben PROLIA. Las fracturas femorales atípicas pueden ocurrir con poco o ningún trauma en las regiones subtrocantéreas y diafisaria del fémur y pueden ser bilaterales. Hallazgos radiológicos específicos caracterizan estos eventos. Las fracturas femorales atípicas también han sido reportados en pacientes con ciertas condiciones de comorbilidad (por ejemplo, la deficiencia de vitamina D, arthiritis reumatoide, hipofosfatasia) y con el uso de ciertos agentes farmacéuticos (por ejemplo, bifosfonatos, glucocorticoides, inhibidores de la bomba de protones). Estos eventos también se han producido sin tratamiento antirresortivo. Durante el tratamiento con PROLIA, los pacientes deben ser notificados para que reporten dolor nuevo o inusual de muslo, cadera o en la ingle. Los pacientes que presentan estos síntomas deben ser evaluados para una fractura de fémur incompleta, y el fémur contralateral también debe ser examinado. La interrupción del tratamiento debe considerarse para cada caso, evaluando el beneficio/riesgo de manera individual. PROLIA contiene el mismo ingrediente activo (denosumab) que se encuentra en Xgeva. Los pacientes que reciben PROLIA no deben recibir Xgeva. Las personas sensibles al látex no deben manipular la tapa de la aguja en la jeringa prellenada de uso único, la cual contiene hule natural seco (un derivado del látex). Efectos sobre la capacidad para conducir y utilizar maquinaria: No se han llevado a cabo estudios sobre el efecto en la capacidad para conducir o utilizar maquinaria pesada en pacientes que reciben denosumab.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: No existen datos adecuados acerca de mujeres embarazadas. PROLIA no se recomienda para uso en mujeres embarazadas. En las exposiciones de ABC hasta 100 veces mayores a la exposición humana (60 mg cada 6 meses), el denosumab no mostró evidencia de discapacidad en la fertilidad en monos cynomolgus. En un estudio en monos cynomolgus que recibieron una dosis de denosumab durante el período equivalente al primera trimestre, con exposiciones de ABC hasta 99 veces mayores que la dosis humana (60 mg cada 6 meses), no hubo evidencia de daño materno o fetal. En este estudio, no se examinaron los ganglios linfáticos del feto. En otro estudio en monos cynomolgus que recibieron una dosis de denosumab durante el embarazo exposiciones de ABC 119 veces mayores que la dosis humana (60 mg cada 6 meses), hubo un incremento de mortinatos y de la mortalidad postnatal; el crecimiento anormal del hueso dio como resultado la reducción de la resistencia ósea, reducción de la hematopoyesis, y mala alineación de los dientes; ausencia de ganglios linfáticos periféricos, y disminución del crecimiento del recién nacido. No hubo evidencia de daño maternal antes del parto; los efectos adversos maternos fueron poco frecuentes durante el parto. El desarrollo de la glándula mamaria materna fue normal. Los estudios en ratones knockout sugieren que la ausencia de RANKL podría interferir con la maduración de la glándula mamaria materna lo que conduce a una insuficiencia en la lactancia posparto. Lactancia: No se conoce si denosumab es excretado en la leche materna. Ya que el denosumab tiene el potencial de provocar reacciones adversas en infantes durante la lactancia, se debe tomar la decisión, ya sea de discontinuar la lactancia o discontinuar el medicamento.
Reacciones secundarias y adversas: Datos de estudios clínicos: Las reacciones adversas se mencionan a continuación por el grupo sistémico corporal de MedDRA y por su frecuencia. Las categorías de frecuencia con base en las tasas de eventos de un año son: Muy común: ≥ 1 en 10. Común: ≥ 1 en 100 y menor a 1 en 10. Poco común: ≥ 1 en 1,000 y < 1 en 100. Rara: ≥ 1 en 10,000 y < 1 en 1,000. Muy rara: < 1/10,000. Dentro de cada grupo de frecuencia y grupo sistémico, los efectos no deseables se presentan en orden de disminución de severidad.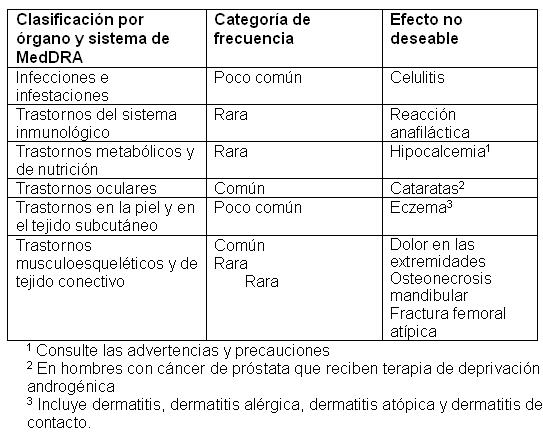
Datos Postcomercialización: Reacciones de Hipersensibilidad: Reacciones de hipersensibilidad, incluyendo rash, urticaria, hinchazón de la cara, eritema y reacciones anafilácticas, han sido reportadas raramente en pacientes que reciben PROLIA. Hipocalcemia severa: Se ha reportado hipocalcemia sintomática severa en pacientes con mayor riesgo de hipocalcemia al recibir PROLIA.
Interacciones medicamentosas y de otro género: PROLIA (60 mg SC) no afectó la farmacocinética de midazolam, que se metaboliza por el citocromo P450 3A4 (CYP3A4), lo que indica que no debería afectar a la farmacocinética de los fármacos metabolizados por esta enzima (consultar propiedades farmacocinéticas). Anormalidades en pruebas de laboratorio: No conocidas.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Carcinogenicidad: No se ha evaluado el potencial carcinogénico del denosumab en estudios a largo plazo con animales. Mutagenicidad: El potencial genotóxico del denosumab no ha sido evaluado. Toxicología reproductiva: Fertilidad: El denosumab no tiene efecto alguno en la fertilidad de las hembras o en los órganos reproductores de los machos en monos en las exposiciones de ABC que fueron de 100 a 150 veces superiores que la exposición en humanos de 60 mg administrados subcutáneamente una vez cada 6 meses. Embarazo: Con exposiciones hasta 100 veces superiores que la exposición humana, el denosumab no mostró evidencia alguna de daño en el feto en los estudios de toxicidad del desarrollo.
Dosis y vía de administración: Administración:La administración debe llevarse a cabo por un individuo que esté capacitado en forma adecuada en técnicas de inyección. Dosis: La dosis recomendada de PROLIA es una inyección subcutánea de 60 mg administrada una vez cada seis meses. Los pacientes deberán recibir complementos de calcio y vitamina D mientras se someten al tratamiento. Poblaciones especiales: Niños: PROLIA no se recomienda en pacientes pediátricos debido a que no se han establecido la seguridad y la efectividad de PROLIA en el grupo de edad pediátrica. Los primates adolescentes que recibieron una dosis de denosumab entre 27 y 150 veces (10 y 50 mg/kg) la exposición clínica basada en el ABC tuvieron placas de crecimiento anormales. En monos cynomolgus recién nacidos expuestos en el útero a denosumab 50 mg/kg, hubo un incremento en la mortalidad postnatal; el crecimiento anormal de los huesos que dio como resultado una reducción de la resistencia ósea, reducción de la hematopoyesis, y mala alineación dental; ausencia de ganglios linfáticos periféricos, y disminución del crecimiento del recién nacido. Después de un período de recuperación desde el nacimiento hasta los 6 meses de edad, los efectos sobre el hueso volvieron a la normalidad; no hubo efectos adversos sobre la erupción de los dientes; y se observó mineralización de mínima a moderada en múltiples tejidos en un animal de recuperación. En estudios con ratas recién nacidas, la inhibición del ligando RANK/RANKL con un constructo de osteoprotegerina ligado a Fc (OPG-Fc) se ha relacionado con la inhibición del crecimiento óseo y la falta de erupción dental. Por lo tanto, el tratamiento con denosumab puede impedir el crecimiento óseo en niños con placas de crecimiento abiertas y puede inhibir la erupción de la dentición. Se observaron cambios fenotípicos similares en un estudio corroborativo en ratas de 2 semanas de edad, a las que se les administro OPG-Fc. Estos cambios fueron parcialmente reversibles en este modelo al descontinuar la administración de inhibidores de RANKL. Ancianos: Con base en los datos se seguridad y eficacia disponibles en ancianos, no se requiere un ajuste en la dosis (consulte Farmacocinética: Poblaciones de pacientes especiales). Insuficiencia renal: Con base en los datos de seguridad y eficacia disponibles en ancianos, no se requiere un ajuste en la dosis en pacientes con insuficiencia renal (consulte Farmacocinética: Poblaciones de pacientes especiales). Los pacientes con insuficiencia renal severa (tasa de aclaramiento de creatinina menor a 30 ml/min) o que reciben diálisis se encuentran en un riesgo mayor de desarrollar hipocalcemia. El consumo adecuado de calcio y vitamina D es importante en los pacientes con insuficiencia renal severa o que reciben diálisis. Insuficiencia hepática: No se han estudiado la seguridad y eficacia de PROLIA en pacientes con insuficiencia hepática.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: No existen datos disponibles de pruebas clínicas acerca de la sobredosis de PROLIA. El denosumab se administro en estudios clínicos utilizando dosis de hasta 180 mg cada 4 semanas (dosis acumulativas de hasta 1080 mg durante 6 meses).
Presentación(es): Caja con 1 jeringa prellenada con 60mg / 1 mL, en envase de burbuja e instructivo.
Recomendaciones sobre almacenamiento: Consérvese en refrigeración entre 2 y 8°C. No congelar. Mantener la jeringa prellenada dentro de la caja para protejerla de la luz. No agitar. Si se saca del refrigerador, PROLIA debe guardarse a una temperatura ambiente controlada (hasta 25°C) en su caja original, y debe usarse dentro de un período de 30 días.
Leyendas de protección: Manténgase fuera del alcance de los niños. Su venta requiere receta médica. Léase instructivo anexo. No se use PROLIA durante el embarazo, ni la lactancia. No se administre a niños menores de 18 años. Literatura exclusiva para médicos. La cubierta de la aguja contiene caucho natural (un derivado del látex).
Nombre y domicilio del laboratorio: Amgen México S.A. de C.V. Tejocotes s/n, lote 3, Agave I, puerta 40 y 41. Col. San Martín Obispo Tepetlixpan, C.P. 54763. Cuautitlán Izcalli, México.
Número de registro del medicamento: 067M2011SSA IV.