RAPIVIR®
GSK
Denominacion genérica: Valaciclovir.
Forma farmacéutica y formulación: Comprimido. Cada comprimido contiene: Clorhidrato de Valaciclovir equivalente a 500 mg de Valaciclovir. Excipiente cbp 1 comprimido.
Indicaciones terapéuticas: RAPIVIR® está indicado para el tratamiento del herpes zóster. Acelera la resolución del dolor: reduce la duración y la proporción de pacientes con dolor asociado al zóster, lo que incluye las neuralgias aguda y post-herpética. RAPIVIR® está indicado para el tratamiento de las infecciones por herpes simple en piel y membranas mucosas, incluyendo herpes genital inicial y recurrente. RAPIVIR® está indicado en el tratamiento del herpes labial (fuegos). RAPIVIR® está indicado para la prevención de herpes recurrente simple que se manifiesta en piel y membranas mucosas, incluyendo herpes genital. RAPIVIR® puede reducir la transmisión de herpes genital cuando se toma como terapia supresora y se combina con prácticas de sexo seguro. RAPIVIR® está indicado para la profilaxis de la infección por citomegalovirus (CMV) y enfermedad posterior al transplante de órganos. La profilaxis para CMV con RAPIVIR® reduce el rechazo al injerto (pacientes con transplante renal), infecciones oportunistas y otras infecciones por virus herpes simple (HVS por sus siglas en inglés) y virus varicela zóster (VVZ por sus siglas en inglés).
Farmacocinética y farmacodinamia: Farmacocinética: Absorción: Después de la administración oral, el valaciclovir es bien absorbido y convertido en forma rápida y casi completa en aciclovir y valina. Probablemente, esta conversión sea mediada por una enzima aislada de hígado humano que se denomina valaciclovir hidrolasa. La biodisponibilidad de aciclovir en 1000 mg de valaciclovir es de 54% y no se reduce por los alimentos. La farmacocinética de RAPIVIR® no es dosis proporcional. La velocidad y extensión de la absorción disminuye cuando se aumenta la dosis, resultando en aumentos menos que proporcionales de la Cmax considerando el rango de dosis terapéuticas y una biodisponibilidad reducida a dosis por encima de 500 mg. Las concentraciones máximas medias de aciclovir son de 10 a 37 micromoles (2.2 a 8.3 microgramos/mL) tras dosis únicas de 250 a 2000mg de valaciclovir en sujetos saludables con funcionamiento renal normal, y ocurren en una mediana de tiempo de 1 a 2 horas post dosis. Las concentraciones plasmáticas máximas de valaciclovir sólo constituyen el 4% de los niveles de aciclovir, ocurren a un tiempo mediano de 30 a 100 min post dosis, y se encuentran en o por debajo de límite de cuantificación tres horas tras la dosificación. Los perfiles de farmacocinética de valaciclovir y aciclovir son semejantes tras dosis únicas y dosis repetidas. El herpes zóster y el herpes simple no modifican significativamente la farmacocinética de valaciclovir y aciclovir tras la administración oral de valaciclovir. Distribución: La unión de aciclovir a las proteínas plasmáticas ocurre en un porcentaje muy bajo (15%). La penetración en el CSF (siglas en Inglés de líquido cefalorraquídeo), determinada mediante el AUC de la relación CSF/plasma, es aproximadamente de 25% para aciclovir y el metabolito 8-hidroxi-aciclovir (8-OH-ACV), y alrededor del 2.5% para el metabolito 9-(carboximetoxi) metilguanina (CMMG) (véase Farmacocinética: Población de Pacientes Especiales). Metabolismo: Después de la administración oral, RAPIVIR es convertido en aciclovir y L-valina por efecto de primer paso intestinal y/o metabolismo hepático. Aciclovir es convertido en una pequeña extensión en los metabolitos 9-(carboximetoxi) metilguanina (CMMG) mediante la alcohol y aldehido deshidrogenasa, y a 8-hidroxi-aciclovir (8-OH-ACV) mediante la aldehido oxidasa. Aproximadamente 88% de la exposición plasmática total combinada es atribuible al aciclovir, 11% a CMMG y 1% a 8-OH-ACV. Ni RAPIVIR® ni aciclovir es metabolizado por las enzimas de la citocromo P450. Eliminación: En pacientes con funcionamiento renal normal, la vida media de eliminación plasmática de aciclovir después de dosis únicas o múltiples es aproximadamente de 3 hrs. Menos del 1% de la dosis administrada de valaciclovir es recuperada en la orina como fármaco sin cambio. Valaciclovir se elimina en orina principalmente como aciclovir (más del 80% de la dosis recuperada) y el metabolito conocido del aciclovir 9-(carboximetoxi) metilguanina. Población de Pacientes Especiales: Insuficiencia renal: La eliminación de aciclovir se correlaciona con la función renal, y la exposición al aciclovir aumentará con el agravamiento de la insuficiencia renal. En pacientes con enfermedad renal terminal, el promedio de la vida media de eliminación de aciclovir después de la administración de RAPIVIR® es aproximadamente de 14 horas, comparado con aproximadamente 3 horas con una función renal normal (véase Dosis y Administración). La exposición al aciclovir y sus metabolitos CMMG y 8-OH-ACV en plasma y líquido cefalorraquideo (CSF por sus siglas en Inglés) se evaluó en situación de equilibrio después de la administración de dosis múltiples de RAPIVIR® en 6 sujetos con función renal normal (depuración promedio de creatinina de 111 mL/min, en un rango de 91-144 mL/min) recibiendo 2000 mg cada 6 horas y en 3 sujetos con insuficiencia renal severa (CLcr 26 mL/min promedio, en un rango de 17-31 mL/min) recibiendo 1500 mg cada 12 horas. Tanto en plasma como en CSF, las concentraciones de aciclovir, CMMG y 8-OH-ACV estuvieron en el rango de 2, 4 y 5-6 veces más altas, respectivamente, en insuficiencia renal severa comparado con la función renal normal. No hubo diferencia en el nivel de penetración al CSF (determinado por el AUC de la relación de CSF/plasma) para aciclovir, CMMG o 8-OH- aciclovir entre las dos poblaciones (véase Farmacocinética: Distribución). Insuficiencia Hepática: Los datos farmacocinéticos indican que la insuficiencia hepática disminuye el índice de conversión de RAPIVIR® en aciclovir pero no la extensión de dicha conversión. La vida media de aciclovir no es afectada. Mujeres embarazadas: En un estudio de la farmacocinética de RAPIVIR® y aciclovir durante embarazo avanzado, la situación de equilibrio AUC de aciclovir diario después de RAPIVIR® 1000 mg diario fue aproximadamente 2 veces más grande que la observada con 1200 mg orales diarios de aciclovir. Para información del paso a la leche maternal véase la sección de Lactancia. Infección con HIV: En pacientes con infección con HIV, las características de disposición y farmacocinética de aciclovir después de la administración oral de dosis única o múltiples de 1000 mg o 2000 mg de RAPIVIR® están inalteradas comparadas con sujetos sanos. Trasplante de órganos: En receptores de trasplante recibiendo 2000 mg de RAPIVIR® 4 veces al día, las concentraciones pico de aciclovir son similares o mayores que aquellas en voluntarios sanos recibiendo las misma dosis. Las AUCs estimadas diarias son apreciablemente mayores. Farmacodinamia: Mecanismo de acción: Valaciclovir, un antiviral, es el éster de L-valina del aciclovir. El aciclovir es un nucleósido análogo de la purina (guanina). Valaciclovir se convierte de manera rápida y casi completa en el hombre a aciclovir y valina, probablemente por la enzima llamada valaciclovir hidrolasa. El aciclovir es un inhibidor específico de virus de herpes con actividad in vitro contra virus de herpes simple (HVS) tipo 1 y tipo 2, virus de varicela zóster (VVZ), citomegalovirus (CMV), Virus de Epstein-Barr (VEB) y virus de herpes humano 6 (VHH-6). El aciclovir inhibe la síntesis de ADN del virus herpético una vez que ha sido fosforilado a la forma activa de trifosfato. La primera etapa de fosforilación requiere la actividad de una enzima específica para el virus. En el caso de HVS, VVZ y VEB esta enzima es la viral timidina cinasa (TK), que sólo está presente en células infectadas por virus. La selectividad se mantiene en CMV con fosforilación, por lo menos parcialmente mediada a través del producto de gen fosfotransferasa de UL97. Este requisito para activación de aciclovir por una enzima específica para el virus explica en gran parte su selectividad. El proceso de fosforilación se completa (conversión de mono a trifosfato) mediante cinasas celulares. El trifosfato de aciclovir inhibe competitivamente la ADN polimerasa viral y la incorporación de los resultados análogos de nucleósido dan lugar a terminación obligada de la cadena, deteniendo así la síntesis de ADN viral y bloqueando por lo tanto la replicación del virus. Efectos farmacodinámicos: La resistencia normalmente se debe a un fenotipo deficiente en timidina cinasa que produce un virus con desventajas profundas en el huésped natural. Con poca frecuencia, se ha descrito reducción de sensibilidad al aciclovir como resultado de alteraciones sutiles en la timidina cinasa o ADN polimerasa viral. La virulencia de estas variantes se asemeja a la de virus de tipo salvaje. El monitoreo extenso de muestras clínicas de HVS y VVZ de pacientes bajo terapia o profilaxia con aciclovir ha revelado que los virus con reducción de sensibilidad al aciclovir son sumamente raros en personas inmunocompetentes, y sólo se observan con muy poca frecuencia en individuos severamente inmunocomprometidos, p. ej., receptores de trasplante de órgano o médula ósea, pacientes bajo quimioterapia por enfermedad maligna y personas infectadas con virus de inmunodeficiencia humana (VIH).
Contraindicaciones: Pacientes con hipersensibilidad a valaciclovir, aciclovir o a alguno de los componentes de la fórmula. Pacientes menores de 12 años de edad.
Precauciones generales: Estado de Hidratación: Se deberá tener cuidado de asegurar el adecuado aporte de líquidos a pacientes que están en riesgo de deshidratación, particularmente los ancianos. Uso en pacientes con afección renal y pacientes de edad avanzada: Aciclovir se elimina por depuración renal, por lo tanto la dosis de valaciclovir debe reducirse en pacientes con afección renal (ver Dosificación y Administración). Es probable que los pacientes de edad avanzada tengan funcionamiento renal reducido y por lo tanto debe tomarse en cuenta la necesidad de una reducción en este grupo de pacientes. Tanto los pacientes de edad avanzada como los que padecen afecciones renales corren mayor riesgo de desarrollar efectos neurológicos secundarios y deben ser monitoreados de manera cercana para evidencia de estos efectos. En los casos reportados, estas reacciones en general fueron reversibles al descontinuar el tratamiento (ver Reacciones Adversas). Empleo de altas dosis de RAPIVIR® en daño hepático y en transplantados de hígado: No hay información disponible sobre el empleo de RAPIVIR® a altas dosis (4 g/día ó más) en pacientes con enfermedad hepática, por lo que se deberá tener precaución si se decide administrar altas dosis de valaciclovir en estos pacientes. No se han llevado a cabo estudios específicos del empleo de valaciclovir en pacientes con transplante de hígado; sin embargo, altas dosis de aciclovir como profilaxis han demostrado disminuir la infección por citomegalovirus y la enfermedad. Uso en herpes genital: la terapia supresora con RAPIVIR® reduce el riesgo de transmisión de esta enfermedad. No cura ni elimina completamente el riesgo de transmisión. Además de la terapia con RAPIVIR® se deben continuar las prácticas de sexo seguro. Efectos sobre la Capacidad para Conducir y Usar Maquinaria: El estatus clínico del paciente y el perfil de eventos adversos de RAPIVIR® deben tenerse en cuenta al considerar la capacidad del paciente para conducir u operar maquinaria. No se han realizado estudios para investigar el efecto de valaciclovir sobre el comportamiento de manejo o la capacidad para operar maquinaria. Además, es imposible predecir un efecto nocivo sobre esas actividades por la farmacología de la sustancia activa.
Restricciones de uso durante el embarazo y la lactancia: Embarazo: Hay datos limitados sobre el uso de RAPIVIR® en el embarazo. RAPIVIR® sólo debe usarse durante el embarazo si los beneficios potenciales del tratamiento exceden el riesgo potencial. Los registros de embarazo tienen documentados resultados del embarazo en mujeres expuestas a RAPIVIR® o a cualquier formulación de ZOVIRAX® (aciclovir, el metabolito activo del valaciclovir); se obtuvieron 111 y 1246 resultados (29 y 756 expuestos durante el primer trimestre del embarazo), respectivamente, de mujeres registradas prospectivamente. Los hallazgos del registro de embarazo de aciclovir no han mostrado aumento en el número de defectos de nacimiento entre las mujeres registradas prospectivamente. Los hallazgos del registro de embarazo de aciclovir no han mostrado aumento en el número de defectos de nacimiento entre las mujeres expuestas a aciclovir en comparación con la población general, y ningún defecto de nacimiento con un patrón único en su género o consistente, que sugiera una causa común. Dado el pequeño número de mujeres incluidas en el registro de embarazo de valaciclovir, no pudieron obtenerse conclusiones confiables y definitivas referentes a la seguridad de valaciclovir en el embarazo. Lactancia: aciclovir es el metabolito activo del valaciclovir que es excretado en la leche materna. Después de la administración de 500 mg de valaciclovir, las concentraciones máximas (Cmax) en la leche materna oscilaron del 0.5 a 2.3 (mediana de 1.4 veces) veces las concentraciones maternas de aciclovir sérico. La relación entre el aciclovir bajo la curva (ABC) en la lecha materna con las concentraciones séricas fue de 1.4 a 2.6 veces (mediana 2.2). La mediana de la concentración de aciclovir en la leche materna fue de 2.24 mg/mL (9.95 mM). Con una dosis materna de 500 mg de valaciclovir dos veces al día, esta dosis expondría al lactante al equivalente a una dosis oral de 0.61 mg/kg/día. La vida media de eliminación del valaciclovir de la leche materna fue similar a la sérica. No se detectó valaciclovir sin alteraciones en el suero materno, leche materna u orina del lactante. En consecuencia, se recomienda precaución si se administra RAPIVIR® a una mujer que está amamantando, sin embargo ZOVIRAX® se emplea para tratar el herpes simple a la dosis intravenosa de 30 mg/kg/día.
Reacciones secundarias y adversas: A continuación se expresan en función del tipo de órganos afectados, de acuerdo a las siguientes categorías por frecuencia: Muy comunes: ≥1 en 10. Comunes: ≥ 1 en 100 y < 1 en 10. No Comunes: ≥1 en 1 000 y < 1 en 100. Raras: ≥1 en 10 000 y < 1 en 1 000. Muy raras: < 1 en 10 000. Se han utilizado los datos de los estudios clínicos para asignar categorías por la frecuencia de aparición de determinados eventos adversos, si existió evidencia de su asociación con el valaciclovir (por ejemplo una diferencia estadísticamente significativa entre quienes recibieron valaciclovir y el placebo). Para todos los demás eventos adversos, se utilizó la información post-comercialización de los reportes espontáneos como base para determinar la categoría por frecuencia. Datos de estudios clínicos: Alteraciones del sistema nervioso central: Comunes: cefalea. Alteraciones gastrointestinales: Común: Náusea. Datos Posteriores a la Comercialización: Alteraciones linfáticas y hematológicas: Muy raras: leucopenia, trombocitopenia. La leucopenia es principalmente reportada en los pacientes inmunocomprometidos. Alteraciones del sistema inmune: Muy raras: anafilaxia. Alteraciones psiquiátricas y del sistema nervioso: Raras: mareos, confusión, alucinaciones, disminución de la conciencia. Muy raras: agitación, temblor, ataxia, disartria, síntomas psicóticos, convulsiones, encefalopatía, coma. Los eventos anteriores son reversibles y usualmente aparecen en pacientes con insuficiencia renal u otras alteraciones predisponentes. En pacientes postransplantados que reciben dosis elevadas de RAPIVIR® (8 g diariamente) para profilaxis de infección por CMV, los eventos neurológicos ocurrieron con mayor frecuencia en comparación con quienes recibieron dosis menores. Alteraciones respiratorias, torácicas y mediastinales: No comunes: disnea. Alteraciones gastrointestinales: Raras: molestias abdominales, vómito, diarrea. Alteraciones hepato-biliares: Muy raras: aumentos reversibles en las pruebas de función hepática. Ocasionalmente descritas como hepatitis. Alteraciones en piel y tejido subcutáneo: No comunes: eritema, incluyendo fotosensibilidad. Raras: prurito. Muy raras: urticaria, angioedema. Alteraciones renales y urinarias: Raras: insuficiencia renal. Muy raras: insuficiencia renal aguda, dolor renal. El dolor renal puede estar asociado con insuficiencia renal. Otras: se han recibido reportes de insuficiencia renal, microangiopatía hemolítica, anemia y trombocitopenia (algunas veces asociados) en pacientes severamente inmunocomprometidos particularmente aquellos con enfermedad avanzada por VIH/SIDA, que reciben altas dosis (8 g al día) de valaciclovir por tiempos prolongados en estudios clínicos. Estos hallazgos también se han observado en pacientes no tratados con valaciclovir con las mismas condiciones subyacentes.
Interacciones medicamentosas y de otro género: No se han identificado interacciones clínicamente significativas. El aciclovir se elimina principalmente sin cambios por la orina, vía secreción tubular activa. Cualquier droga que se administre concomitantemente y que compita con este modo de eliminación puede incrementar las condiciones plasmáticas de aciclovir, después de la administración de RAPIVIR®. Después de administrar un gramo de RAPIVIR®, la cimetidina y el probenecid aumentan el área bajo la curva de aciclovir por este mecanismo y reducen el aclaramiento renal de aciclovir. Sin embargo no se requiere ajuste de la dosis en este rango en virtud del amplio índice terapéutico de aciclovir. Los pacientes que reciben dosis elevadas de RAPIVIR® (4 g ó más al día), se recomienda cautela si se administran otros medicamentos que compiten con el aciclovir en su eliminación, por el potencial aumento de niveles plasmáticos de uno o ambos metabolitos de los diversos medicamentos o drogas. Se ha demostrado un aumento en el área bajo la curva de aciclovir y del metabolito inactivo mofetil de micofenolato, un agente inmunosupresor utilizado en pacientes transplantados, cuando aciclovir oral y micofenolato mofetil son coadministrados. Se requiere también cuidado (con monitoreo de la función renal) si se administran dosis más altas de RAPIVIR® (4 g ó más al día) con drogas que afecten otros aspectos de la función renal (ciclosporina y tacrolimo).
Alteraciones en los resultados de pruebas de laboratorio: No se han reportado a la fecha.
Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Carcinogénesis: Valaciclovir no demostró ser carcinogénico en los ensayos preclínicos realizados en ratones y ratas. Mutagénesis: Los resultados de las pruebas de mutagenicidad in vitro e in vivo indicaron que es poco probable que el valaciclovir provoque un riesgo genético en el humano. Teratogénesis: Valaciclovir no fue teratogénico en rata ni conejo. Valaciclovir se metaboliza casi en su totalidad a aciclovir. La administración subcutánea de aciclovir en pruebas de aceptación internacional no ocasionó efectos teratogénicos en rata o conejo. En estudios adicionales en rata, se observaron anormalidades fetales a dosis subcutáneas que produjeron niveles plasmáticos de 100 microgramos/mL y toxicidad materna. Fertilidad: El valaciclovir administrado por vía oral, no afectó la fertilidad en ratas hembras o machos. Se ha observado atrofia testicular y espermatogénesis con dosis parenterales altas en ratas y perros. En estudios con animales, RAPIVIR® no afecta la fertilidad. Sin embargo, dosis altas parenterales de aciclovir causó efectos testiculares en ratas y perros (véase Datos de Seguridad Preclínica). No se realizaron estudios de fertilidad en humanos con RAPIVIR®, pero no se reportaron cambios en la cuenta la motilidad o en la morfología espermática, en 20 pacientes después de 6 meses de tratamiento diario con 400 mg a 1 g de aciclovir.
Dosis y via de administración: Vía de administración: Oral. Adultos: Tratamiento del Herpes zóster: La dosis en adultos es de 1000 mg de RAPIVIR® tres veces al día durante 7 días. Tratamiento de Infecciones por Herpes simple: Adultos y adolescentes inmunocompetentes (mayores de 12 años): La dosis en adultos es de 500 mg de RAPIVIR® dos veces al día. Para episodios de recurrencia, el tratamiento debe ser de 3 a 5 días. Para episodios de primera vez, los cuales pueden ser más severos, el tratamiento se puede prolongar de 5 hasta 10 días. La dosis se debe iniciar lo antes posible. Para episodios recurrentes de herpes simple, esto debe tener lugar de manera ideal en el periodo prodrómico o inmediatamente que aparezcan los primeros signos o síntomas, RAPIVIR® puede prevenir el desarrollo de lesiones cuando se toma al ocurrir los primeros signos y síntomas de una recaída de HVS. Alternativamente para herpes labial (fuegos) RAPIVIR® debe tomarse a razón de 2 g dos veces al día durante un día constituye un tratamiento eficaz. La segunda dosis debe ser tomada hasta 12 hrs. (no antes de 6 hrs.) después de la primera dosis. Cuando se utiliza este régimen el tratamiento no debe exceder más de un día, ya que no se ha visto un beneficio terapéutico adicional. El tratamiento debe iniciarse lo antes posible, precisamente en el episodio prodrómico (al presentarse comezón, sensación de quemadura y hormigueo). Prevención (supresión) de las infecciones recurrentes herpes simple: Adultos y adolescentes inmunocompetentes (mayores de 12 años): En pacientes adultos inmunocompetentes, la dosis es de 500 mg de RAPIVIR® una vez al dia. En algunos pacientes con recurrencias muy frecuentes (por ejemplo 10 o más al año) pueden obtener un beneficio adicional si toman una dosis diaria de 500 mg dividida en 250 mg dos veces al día. Adultos inmunocomprometidos: Para pacientes inmunocomprometidos la dosis es de 500 mg dos veces al día. Reducción de la transmisión del herpes genital: En adultos heterosexuales inmunocompetentes con menos de nueve episodios recurrentes al año, 500 mg de RAPIVIR® una vez al día, es la dosis recomendada a la pareja sexual. No existen datos sobre la reducción en la transmisión en otro tipo de pacientes. Profilaxis para la infección por citomegalovirus (CMV): Dosis en adultos y adolescentes (desde 12 años de edad): La dosis de RAPIVIR® es de 2 gramos cuatro veces al día, y debe iniciarse tan pronto sea posible posterior al transplante. Esto se deberá reducir de acuerdo al aclaramiento de creatinina. La duración del tratamiento será generalmente de 90 días, pero puede ser necesario prolongarla en pacientes de alto riesgo. Dosis en Insuficiencia Renal: Se debe tener cautela al administrar valaciclovir a pacientes con funcionamiento renal afectado. Es preciso mantener hidratación adecuada. La dosis de RAPIVIR® debe reducirse en pacientes con funcionamiento renal significativamente afectado como se muestra en la siguiente tabla: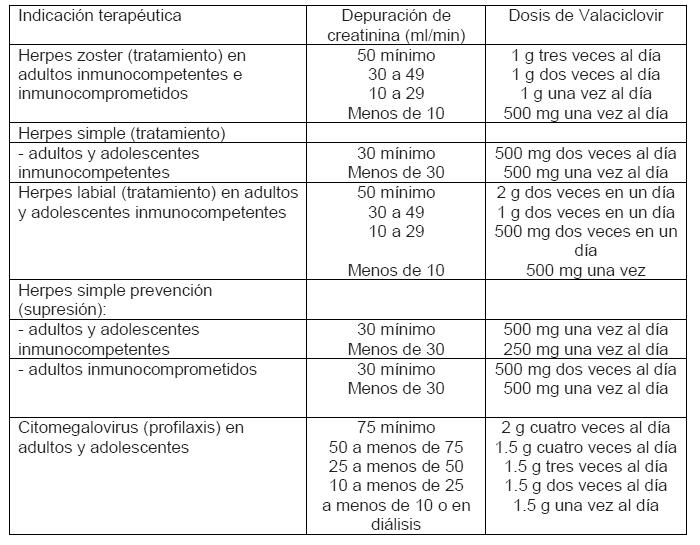
En los pacientes sometidos a hemodiálisis intermitente, la dosis de RAPIVIR® recomendada es la que corresponde a los casos con depuración de creatinina < 15 mL/minuto y deberá ser administrada después de la hemodiálisis. Debe monitorearse con frecuencia la depuración de creatinina, en particular en periodos en que el funcionamiento renal esté sufriendo modificaciones rápidas, p. ej., inmediatamente después del trasplante o del proceso de injerto. La dosis de RAPIVIR® debe ajustarse en consecuencia. Dosis en insuficiencia hepática: Estudios con 1 g de RAPIVIR®, muestran que no se requiere modificación de la dosis en pacientes con cirrosis leve ó moderada (funcionamiento sintético, hepático afectado y evidencia de derivación portal-sistémica) no requieren ajuste de la dosis, sin embargo la experiencia clínica es limitada. Para dosis mayores (4g al día ó más) ver las Precauciones generales y advertencias. Dosis en niños: No hay datos sobre el uso de RAPIVIR®. Dosis en Ancianos: No se requiere de modificaciones en la dosis a menos que la función renal se encuentre deteriorada de manera significativa. Se debe mantener una hidratación adecuada de los pacientes.
Manifestaciones y manejo de la sobredosificación o ingesta accidental: Las dosis repetidas (accidentales) se valaciclovir durante varios días se han asociado a efectos gastrointestinales (náusea y vómito) y neurológicos (cefalea y confusión). La sobredosis de aciclovir intravenoso resultó en elevaciones de la creatinina sérica y la aparición subsecuente de insuficiencia renal. Los efectos neurológicos incluyendo la confusión, alucinaciones, agitación, convulsiones y coma, se han descrito en asociación a dosis elevadas por vía IV. Tratamiento de la sobredosis: se deben buscar signos de toxicidad mediante la observación cuidadosa de los pacientes. La hemodiálisis aumenta significativamente la eliminación del aciclovir sanguíneo y en consecuencia puede considerarse una opción de tratamiento en los casos de sobredosificación sintomática.
Presentaciones: Caja con 6, 10, 15, 18, 30, 36 ó 42 comprimidos recubiertos en envase de burbuja.
Recomendaciones sobre almacenamiento: Consérvese a no más de 30°C y en lugar seco. Protéjase de la luz.
Leyendas de protección: Su venta requiere receta médica. No se deje al alcance de los niños. Literatura exclusiva para médicos. Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx.
Nombre y domicilio del laboratorio: GlaxoSmithKline México, S.A. de C.V. Calzada México Xochimilco No. 4900, Colonia San Lorenzo Huipulco, C.P. 14370, Deleg. Tlalpan, D.F. México.
Número de registro del medicamento: 381M95 SSA IV
Clave de IPPA: GDS22-IPI04-18-JUNIO-2012