RELVARE®
GSK
Denominacion genérica: Furoato de Fluticasona/Vilanterol.
Forma farmacéutica y formulación: Cada dosis pre-dispensada contiene ya sea 100/25 microgramos o 200/25 microgramos de furoato de fluticasona /vilanterol (como trifenatato). Cada inhalación única de furoato de fluticasona/vilanterol proporciona una dosis entregada de 92/22 microgramos de furoato de fluticasona/vilanterol o 184/22 microgramos de furoato de fluticasona/vilanterol. Polvo para inhalación: Cada dosis contiene: Furoato de Fluticasona 92 mcg y 184 mcg. Trifenatato de Vilanterol 22 mcg y 22 mcg. Vehículo/Excipiente cbp 12.5 mg.
Indicaciones terapéuticas: Asma: RELVARE® está indicado para el tratamiento de mantenimiento del asma. Epoc: RELVARE® está indicado para el tratamiento de mantenimiento de la obstrucción de vías aéreas en pacientes con enfermedad pulmonar obstructiva crónica (EPOC), incluyendo bronquitis crónica y/o enfisema, y para reducir las exacerbaciones de EPOC en pacientes con historia de exacerbaciones.
Farmacocinética y farmacodinamia: Farmacodinamia: Mecanismo de acción: Furoato de fluticasona y vilanterol representan dos clases de medicamentos (un corticoesteroide sintético y un agonista de los receptores beta2 de acción prolongada). Efectos farmacodinámicos: Furoato de Fluticasona: Furoato de Fluticasona es un corticoesteroide trifluorinado sintético con una actividad antiinflamatoria potente. Se desconoce el mecanismo preciso a través del cual el furoato de fluticasona afecta los síntomas de asma y EPOC. Se ha demostrado que los corticosteroides tienen un amplio rango de acciones sobre varios tipos celulares (ej., eosinófilos, macrófagos, linfocitos) y mediadores (ej., citocinas y quimiocinas involucradas en inflamación). Vilanterol Trifenatato: Vilanterol Trifenatato es un agonista beta2adrenérgico selectivo y de larga duración (LABA). Los efectos farmacológicos de los fármacos agonistas de los adrenoreceptores beta2, incluyendo vilanterol trifenatato, son atribuibles al menos en parte a la estimulación de la adenilato ciclasa intracelular, la enzima que cataliza la conversión de adenosin trifosfato (ATP) a 3',5'-adenosin monofosfato cíclico (AMP cíclico). Los niveles aumentados de AMP cíclico ocasionan relajación del músculo liso bronquial e inhibición de la liberación de los mediadores de hipersensibilidad inmediata de las células, especialmente de los mastocitos. Ocurren interacciones moleculares entre los corticoesteroides y los LABA, mientras que los esteroides activan al gen del receptor beta2, aumentando el número y sensibilidad de los receptores; los LABA activan al receptor de glucocorticoides para la activación dependiente de esteroides y la potenciación de la translocación nuclear en la célula. Estas interacciones sinérgicas se reflejan en el aumento de la actividad antiinflamatoria, la cual ha sido demostrada in vitro e in vivo en un amplio rango de células inflamatorias relevantes para la fisiopatología del asma y de EPOC. En células mononucleares en sangre periférica de sujetos con EPOC, se observa un mayor efecto anti-inflamatorio en presencia de la combinación de furoato de fluticasona/vilanterol comparado con furoato de fluticasona sola a concentraciones logradas con las dosis clínicas. Farmacocinética: Absorción: La biodisponibilidad absoluta del furoato de fluticasona y vilanterol cuando se administraron mediante inhalación en forma de RELVARE®, fue en promedio de 15.2% y 27.3%, respectivamente. La biodisponibilidad oral de Furoato de Fluticasona y Vilanterol fue baja, en promedio, 1.26% y < 2%, respectivamente. Debido a esta biodisponibilidad oral baja, la exposición sistémica de furoato de fluticasona y vilanterol después de la administración inhalada se debe principalmente a la absorción de la porción inhalada de la dosis administrada al pulmón. Distribución: Después de la dosificación intravenosa, tanto furoato de fluticasona como vilanterol se distribuyen ampliamente con volúmenes de distribución promedio en estado de equilibrio de 661 L y 165 L, respectivamente. Tanto el furoato de fluticasona como vilanterol presentan una asociación baja con los eritrocitos. La unión a proteínas plasmáticas in vitro en plasma humano de furoato de fluticasona y vilanterol fue alta, en promedio > 99.6% y 93.9%, respectivamente. No se observó una disminución en la extensión de la unión a proteínas plasmáticas in vitro en sujetos con insuficiencia renal o hepática. Furoato de fluticasona y vilanterol son sustratos de la P-gp, sin embargo, se considera poco probable que la administración concomitante de furoato de fluticasona/vilanterol con inhibidores de la P-gp altere la exposición sistémica a furoato de fluticasona o vilanterol, ya que ambos son moléculas que se absorben de forma adecuada. Metabolismo: En base a los datos in vitro, las principales vías de metabolismo tanto de furoato de fluticasona como de vilanterol en humanos, están mediadas principalmente por el CYP3A4. Furoato de fluticasona se metaboliza principalmente mediante la hidrólisis del grupo S-fluorometil carbotioato a metabolitos con actividad de corticoesteroide significativamente disminuida. Vilanterol se metaboliza principalmente mediante la O-dealquilación a un rango de metabolitos con actividad agonista 1 y 2 significativamente disminuida. Se realizó un estudio de dosis repetidas de interacción farmacológica con CYP3A4, en sujetos sanos, con la combinación de furoato de fluticasona/vilanterol (200/25) y el inhibidor potente del CYP3A4, ketoconazol (400mg). La coadministración aumentó la media del AUC(0-24) y la Cmax de furoato de fluticasona en 36% y 33%, respectivamente. El aumento en la exposición a furoato de fluticasona se asoció con una reducción de 27% en la media ponderada a 0-24 h del cortisol sérico. La coadministración aumentó la media del AUC(0-t) y la Cmax de Vilanterol en 65% y 22%, respectivamente. El aumento en la exposición de Vilanterol no se asoció con un aumento de los efectos relacionados con los agonistas beta sobre el ritmo cardiaco, el potasio en sangre o el intervalo QTcF. Eliminación: Después de la administración oral, furoato de fluticasona fue eliminado en humanos principalmente mediante metabolismo, y los metabolitos se excretaron casi exclusivamente en heces, con < 1% de la dosis radioactiva recuperada en orina. La vida media de eliminación plasmática aparente de furoato de fluticasona después de la administración inhalada de furoato de fluticasona/vilanterol fue, en promedio, de 24 horas. Después de la administración oral, vilanterol fue eliminado en humanos principalmente mediante metabolismo, seguido por la excreción de los metabolitos en orina y heces en aproximadamente 70% y 30% de la dosis radioactiva, respectivamente. La vida media plasmática de eliminación aparente de vilanterol después de la administración inhalada de furoato de fluticasona/vilanterol fue, en promedio, de 2.5 horas. Poblaciones Especiales de Pacientes: Se realizaron meta análisis PK poblacionales de furoato de fluticasona y vilanterol en estudios de fase III en sujetos con asma o EPOC. Se evaluó el impacto de las co variables demográficas (edad, género, peso BMI, grupo racial, etnicidad) sobre la farmacocinética de furoato de fluticasona y vilanterol, como parte del análisis farmacocinético poblacional. Raza: En sujetos con asma o EPOC, los cálculos del AUC(0-24) de furoato de fluticasona en sujetos del Este de Asia, japoneses y del Sudeste Asiático (12-14% de los sujetos), fueron hasta 53% más altos en promedio en comparación con sujetos caucásicos. Sin embargo, no se observó evidencia de que la exposición sistémica más alta en estas poblaciones se asociara con un efecto mayor sobre la excreción de cortisol urinario en 24 horas. No se observó un efecto de la raza sobre los cálculos de los parámetros farmacocinéticos en sujetos con EPOC. En promedio, se calcula que la Cmax de vilanterol es de 220 a 287% más alta, y el AUC(0-24) es comparable en sujetos con ascendencia asiática en comparación con sujetos de otros grupos raciales. Sin embargo, no se observó evidencia de que esta Cmax más alta de vilanterol resultara en efectos clínicamente significativos sobre el ritmo cardiaco. Niños: En adolescentes (12 años o mayores), no existen recomendaciones sobre modificaciones de la dosis. No se ha estudiado la farmacocinética de furoato de fluticasona/vilanterol en pacientes menores de 12 años de edad. Aún no se han establecido la seguridad y eficacia de furoato de fluticasona/vilanterol en niños menores de 12 años. Ancianos: Se determinaron los efectos de la edad sobre la farmacocinética de furoato de fluticasona y vilanterol en estudios de fase III en EPOC y asma. No se observó evidencia de que la edad (12 - 84) afecte la PK de furoato de fluticasona y vilanterol en sujetos con asma. No se observó evidencia de que la edad afecte la PK de furoato de fluticasona en sujetos con EPOC, mientras que se observó un aumento (37%) en el AUC (0-24) de vilanterol en el rango de edad observado de 41 a 84 años. En sujetos ancianos (84 años de edad) con peso corporal bajo (35 kg), se predice que el AUC (0-24) de vilanterol será 35% mayor en comparación con el cálculo poblacional (sujeto con EPOC de 60 años con un peso corporal de 70 kg), mientras que la Cmax se mantuvo sin cambios. Es poco probable que estas diferencias sean clínicamente relevantes. Insuficiencia Renal: Un estudio de farmacología clínica realizado con furoato de fluticasona/vilanterol, mostró que la insuficiencia renal severa (depuración de creatinina < 30mL/min), no ocasionó una exposición significativamente mayor a furoato de fluticasona o vilanterol, ni efectos sistémicos más marcados de los corticoesteroides o de los agonistas beta2 en comparación con sujetos sanos. No se requiere ajustar la dosis en pacientes con insuficiencia renal. No se han estudiado los efectos de la hemodiálisis. Insuficiencia Hepática: Después de la dosificación repetida de furoato de fluticasona/vilanterol durante 7 días, se observó un aumento en la exposición sistémica a furoato de fluticasona (hasta de tres veces, medido mediante el AUC(0-24)) en sujetos con insuficiencia hepática, en comparación con sujetos sanos (Child-Pugh A, B o C). El aumento en la exposición sistémica a furoato de fluticasona en sujetos con insuficiencia hepática moderada (furoato de fluticasona/vilanterol 200/25 microgramos), se asoció con una disminución promedio de 34% del cortisol sérico en comparación con sujetos sanos (Child-Pugh B). En sujetos con insuficiencia hepática grave (Child-Pugh C) que recibieron una dosis más baja de 100/12.5 microgramos no hubo una disminución en el cortisol sérico. Para pacientes con insuficiencia hepática moderada o grave la dosis máxima es de 100/25 microgramos (véase Dosis y Administración). Después de la dosificación repetida de furoato de fluticasona/vilanterol durante 7 días, no se observó un aumento significativo en la exposición sistémica a vilanterol (Cmax y AUC) en sujetos con insuficiencia hepática leve, moderada o severa (Child-Pugh A, B o C). No se observaron efectos clínicamente relevantes de la combinación de furoato de fluticasona/vilanterol sobre los efectos beta adrenérgicos sistémicos (ritmo cardiaco o potasio sérico) en sujetos con insuficiencia hepática leve o moderada (vilanterol, 25 microgramos) o con insuficiencia hepática severa (vilanterol, 12.5 microgramos), en comparación con sujetos sanos. Género, Peso e IMC: No se observó evidencia de que el género, el peso o el IMC afecten la farmacocinética de furoato de fluticasona, en base a un análisis farmacocinético poblacional de los datos de fase III en 1213 sujetos con asma (712 mujeres) y 1225 sujetos con EPOC (392 mujeres). No se observó evidencia de que el género, el peso o el IMC afecten la farmacocinética de vilanterol, en base a un análisis farmacocinético poblacional en 856 sujetos con asma (500 mujeres) y 1091 sujetos con EPOC (340 mujeres). No es necesario ajustar la dosis en base al género, el peso o el índice de masa corporal (IMC). Estudios clínicos: Estudios clínicos con RELVARE®: Asma: Se han evaluado la seguridad y eficacia de furoato de fluticasona (FF) y vilanterol (VI) en el tratamiento del asma, en 3 estudios clínicos aleatorizados, doble ciego, de entre 12 a 76 semanas de duración (HZA106827, HZA106829 y HZA106837), involucrando 3,210 pacientes de 12 años de edad y mayores con asma persistente. Todos los sujetos estaban utilizando un ICS (Corticoesteroide Inhalado) con o sin un LABA, al menos durante 12 semanas antes de la visita 1. En HZA106837, todos los pacientes habían presentado al menos una exacerbación que requirió tratamiento con corticosteroides orales en el año previo a la visita 1. Los resultados de HZA106827 y HZA106829 se muestran en la siguiente tabla: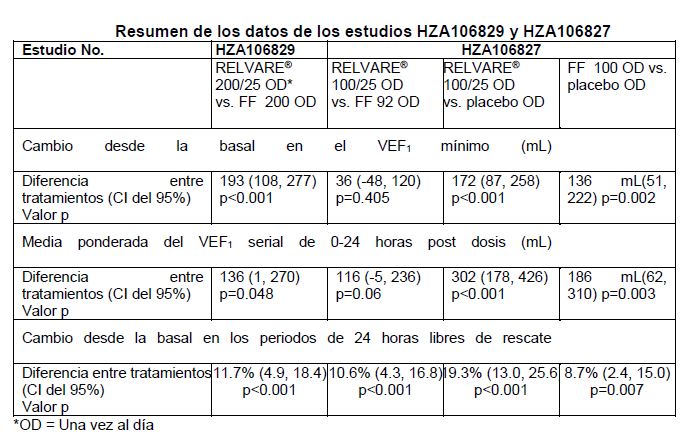
HZA106837 tuvo una duración variable del tratamiento (de un mínimo de 24 semanas a un máximo de 76 semanas, y la mayoría de los pacientes fueron tratados al menos durante 52 semanas) y comparó RELVARE® 92/22 microgramos [N=1009] con FF 92 microgramos [N=1010]. El criterio de valoración primario fue el tiempo hasta la primera exacerbación severa de asma (una exacerbación severa de asma se definió como un deterioro del asma que requirió el uso de corticoesteroides sistémicos u hospitalización del paciente o una visita a urgencias). El riesgo de experimentar una exacerbación severa de asma en pacientes que recibieron RELVARE® 92/22, disminuyó 20% en comparación con FF 92 solo (cociente de riesgo 0.795, p=0.036 IC del 95% (0.642, 0.985)). La tasa de exacerbaciones severas de asma por paciente por año, fue de 0.19 en el grupo de FF 92, y de 0.14 en el grupo de RELVARE® 92/22. La relación de la tasa de exacerbaciones para RELVARE® 92/22 versus FF 92, fue de 0.755 (IC del 95% 0.603, 0.945). Esto representa una disminución de 25% en la tasa de exacerbaciones severas de asma en sujetos tratados con RELVARE® 92/22 en comparación con FF 92 (p=0.014). El efecto broncodilatador en 24 horas de RELVARE® se mantuvo durante un periodo de tratamiento de un año, sin evidencia de pérdida de eficacia (sin taquifilaxia). RELVARE® 92/22 microgramos, demostró de forma consistente, mejorías de 83 mL a 95 mL en el VEF1 (volumen espiratorio forzado) mínimo en las Semanas 12, 36 y 52, y en el Criterio de Valoración en comparación con FF 92 (p < 0.001 IC del 95% 52, 126mL en el Criterio de Valoración). Cuarenta y cuatro por ciento de los pacientes en el grupo de RELVARE® 92/22, estaban bien controlados (ACQ7 ≤0.75) al final del tratamiento, en comparación con 36% de los sujetos en el grupo de FF 92 (p < 0.001 IC del 95% 1.23, 1.82). Enfermedad Pulmonar Obstructiva Crónica: Se ha evaluado la eficacia de furoato de fluticasona y vilanterol en el tratamiento de pacientes con EPOC, en dos estudios de 6 meses (HZC112206, HZC112207), en dos estudios controlados aleatorizados de un año (HZC102970, HZC102871) y un estudio de largo plazo (SUMMIT) en pacientes con diagnóstico clínico de EPOC. Estudios de seis meses: HZC112206 y HZC112207 fueron estudios aleatorizados de 24 semanas, doble ciego, controlados con placebo, de grupos paralelos, que compararon el efecto de la combinación con vilanterol y FF solo y placebo. HZC112206 evaluó la eficacia de RELVARE® 46/22 microgramos [n=206] y RELVARE® 92/22 microgramos [n=206]) en comparación con FF (92 microgramos [n=206]) y vilanterol (22 microgramos [n=205]) y placebo (n = 207), todos administrados una vez al día. HZC112207 evaluó la eficacia de RELVARE® 92/22 microgramos [n=204] y RELVARE® 184/22 [n=205]) en comparación con FF (92 microgramos [n=204] y 184 microgramos [n=203]) y vilanterol (22 microgramos [n=203]) y placebo (n = 205), todos administrados una vez al día. Los criterios de valoración co primarios en ambos estudios, fueron la media ponderada del VEF1 desde cero a 4 horas post-dosis, y el cambio desde la basal en el VEF1 mínimo pre dosis hasta el final del estudio. En un análisis integrado de ambos estudios, RELVARE® 92/22 microgramos mostró mejorías clínicamente significativas de la función pulmonar. En el punto de tiempo de 24 semanas, RELVARE® 92/22 microgramos y vilanterol, aumentaron el VEF1 mínimo 129 mL (IC del 95% 91, 167mL, p < 0.001) y 83mL (IC del 95% 46, 121mL, p < 0.001) respectivamente, en comparación con placebo. RELVARE® 92/22 microgramos aumentó el VEF1 mínimo 46 ml en comparación con Vilanterol (IC del 95% 8, 83mL, p= 0.017). En el punto de tiempo de 24 semanas, RELVARE® 92/22 microgramos y Vilanterol, presentaron una media ponderada del VEF1 de 0-4 horas más alta, de 193 mL (IC del 95% 156, 230mL, p < 0.001) y 145 mL (IC del 95% 108, 181mL, p < 0.001), respectivamente, en comparación con placebo. La diferencia en la media ponderada del VEF1 en 0-4 horas entre los grupos de furoato de fluticasona/vilanterol 92/22 y vilanterol, fue de 48 (95 IC 12, 84 mL, p= 0.009). Estudios de 12 meses: Los estudios HZC102970 y HZC102871 fueron estudios aleatorizados, de 52 semanas, doble ciego, de grupos paralelos, comparando la eficacia y seguridad de RELVARE® 184/22 microgramos, RELVARE® 92/22 microgramos, furoato de fluticasona/vilanterol 50/25 microgramos y vilanterol 22 microgramos, todos administrados una vez al día. El criterio de valoración primario fue la disminución en la tasa anual de exacerbaciones moderadas y severas en sujetos con EPOC. Los resultados de ambos estudios mostraron que el tratamiento con RELVARE® 92/22 microgramos una vez al día, resultó en una disminución de 27% en la tasa anual de exacerbaciones moderadas a severas de EPOC en comparación con vilanterol (IC del 95%:16, 37mL (p≤0.001). Comparado con vilanterol, con furoato de fluticasona /vilanterol 100/25 microgramos una vez al día también se observaron reducciones en el riesgo del tiempo para la primera exacerbación moderada o grave y el índice de exacerbaciones requiriendo el uso de corticoesteroides. En un análisis agrupado de HZC102970 y HZC102871 en la Semana 52, el grupo de RELVARE® 92/22 microgramos, mostró una mejoría mayor del VEF1 mínimo en comparación con el grupo de vilanterol 22 microgramos (diferencia de 42 mL en el cambio medio ajustado desde la basal; IC del 95%: 19, 64mL, p < 0.001). Estudio de largo plazo: SUMMIT fue un estudio multi-céntrico, randomizado, doble-ciego que evaluó el efecto en la sobrevida de RELVARE® 100/25 microgramos comparado con placebo en 16,568 sujetos. Los sujetos fueron tratados hasta 4 años (promedio 1.7 años) tanto con RELVARE® 100/25 microgramos, furoato de fluticasona 100 microgramos, vilanterol 25 microgramos, o placebo. Todos los sujetos tenían EPOC con limitación moderada de flujo de aire (≥50% y ≤70% del FEV1 predicho) y una historia de, o un riesgo aumentado de enfermedad cardiovascular. La sobrevida con RELVARE® no mejoró significativamente comparado con placebo (HR 0.878; 95% IC: 0.739, 1.042; p=0.137), FF (HR 0.964; 95% IC: 0.808, 1.149; p=0.681) o VI (HR 0.912; 95% IC: 0.767, 1.085; p=0.299). La mortalidad por cualquier causa fue: furoato de fluticasona/vilanterol, 6.0%; placebo, 6.7%; furoato de fluticasona, 6.1%; vilanterol, 6.4%). RELVARE® redujo el índice de declive de la función pulmonar medida mediante el FEV1, en 8 mL/año comparado con placebo (95% IC: 1, 15; p=0.019). No hubo impacto (0 mL/year; 95% IC: -6, 7; p=0.913) en el índice del declive por RELVARE® comparado con furoato de fluticasona; hubo una diferencia de 10 mL/año por RELVARE® comparado con vilanterol (95% IC: 3, 16; p=0.004). El índice de declive promedio en el FEV1 fue: RELVARE®, 38 mL/año; placebo, 46 mL/año; furoato de fluticasona, 38 mL/año; vilanterol, 47 mL/año. El riesgo de un evento cardiovascular compuesto (muerte de causa cardiovascular, infarto al miocardio, ictus, angina inestable, o crisis isquémica transitoria, durante el tratamiento) con RELVARE® no fue significativamente más bajo que con placebo (HR 0.926; 95% IC: 0.750, 1.143; p=0.475), FF (HR 1.033; 95% IC: 0.834, 1.281; p=0.763) o VI (HR 0.938; 95% IC: 0.761, 1.155; p=0.545). La incidencia de eventos cardiovasculares compuestos fue: RELVARE®, 4.2%; placebo, 4.2%; furoato de fluticasona, 3.9%; vilanterol 4.4%. RELVARE® demostró un mayor cambio promedio a partir de la basal en el FEV1 post-broncodilatador al Día 360 comparado con placebo (89 mL; 95% IC: 76, 102; p < 0.001), FF (40 mL; 95% IC: 27, 53; p < 0.001), y VI (26 mL; 95% IC: 13, 39; p < 0.001). El cambio promedio ajustado a partir de la basal fue: RELVARE® l 50 mL, placebo, -39 mL; furoato de fluticasona, 9 mL; vilanterol, 24 mL. RELVARE® redujo el índice anual de exacerbaciones moderadas o graves en 29% (95% IC: 22, 35; p < 0.001) comparado con placebo, en 19% comparado con FF (95% IC: 12, 26; p < 0.001) y en 21% comparado con VI (95% IC: 14, 28; p < 0.001). El índice anual de exacerbaciones moderadas o graves fue 0.25 para RELVARE®, 0.35 para placebo, 0.31 para furoato de fluticasona, y 0.31 para vilanterol. RELVARE® redujo el índice anual de exacerbaciones graves (P.Ej. requiriendo hospitalización) en 27% (95% IC: 13, 39; p < 0.001) comparado con placebo, en 11% comparado con FF (95% IC: -6, 25; p=0.204) y en 9% comparado con VI (95% IC: -8, 24; p=0.282). El índice anual de exacerbaciones requiriendo hospitalización fue 0.05 para RELVARE®, 0.07 para placebo, 0.06 para furoato de fluticasona, y 0.06 para vilanterol.
Contraindicaciones: RELVARE® está contraindicado en pacientes con alergia severa la proteína de la leche, o quienes hayan demostrado hipersensibilidad a furoato de fluticasona, vilanterol o a cualquiera de los excipientes. Pacientes menores de 12 años.
Precauciones generales: RELVARE® no debe utilizarse para tratar los síntomas de asma agudo o una exacerbación aguda de EPOC, para los cuales se requiere un broncodilatador de acción rápida. Si se aumenta el uso de los broncodilatadores de acción rápida para aliviar los síntomas, esto indica un deterioro del control del asma, por lo que el paciente debe ser revisado por un médico. Los pacientes no deben suspender el tratamiento con RELVARE® por asma o EPOC sin supervisión del médico, ya que los síntomas pueden recurrir después de la suspensión. Pueden ocurrir eventos adversos y exacerbaciones relacionados con el asma durante el tratamiento con RELVARE®. Debe pedirse a los pacientes que continúen el tratamiento pero que acudan al médico si los síntomas de asma se mantienen sin control o si empeoran después de iniciar RELVARE®. Broncoespamo paradójico: Al igual que con otros tratamientos inhalados, puede ocurrir broncoespasmo paradójico, con un aumento inmediato de las sibilancias después de la dosificación. Esto debe tratarse de inmediato con un broncodilatador inhalado de acción rápida RELVARE® debe suspenderse de inmediato, el paciente debe ser evaluado y debe iniciarse un tratamiento alternativo si es necesario. Efectos Cardiovasculares: Pueden observarse efectos cardiovasculares, como arritmias cardiacas, por ejemplo, taquicardia supraventricular y extrasístoles, con los fármacos simpaticomiméticos, incluyendo RELVARE®. En un estudio controlado con placebo en sujetos con historia de, o un aumento en el riesgo de enfermedad cardiovascular, no hubo aumento en el riesgo de eventos cardiovasculares, eventos cardiovasculares graves, o adjudicación de muertes de causa cardiovascular en pacientes recibiendo furoato de fluticasona /vilanterol comparado con placebo (véase Reacciones Adversas). Sin embargo RELVARE® debe utilizarse con precaución en pacientes con enfermedad cardiovascular severa. Pacientes con insuficiencia hepática: Para los pacientes con insuficiencia hepática moderada o grave, la dosis que debe usarse es 100/25 microgramos y deben ser monitoreados para reacciones adversas sístémicos relacionados con corticosteroides (véase Farmacocinética). Efectos sistémicos de corticosteroides: Pueden ocurrir efectos sistémicos con cualquier corticosteroide inhalado, particularmente con dosis altas prescritas por periodos prolongados. Es menos probable que estos efectos ocurran en comparación con los corticosteroides orales. Los posibles efectos sistémicos incluyen supresión del eje HPA, disminución de la densidad mineral ósea, retardo del crecimiento en niños y adolescentes, catarata y glaucoma. Al igual que con todos los medicamentos que contienen corticosteroides, RELVARE® debe administrarse con precaución en pacientes con tuberculosis pulmonar o en pacientes con infecciones crónicas o no tratadas. Neumonía: Se ha observado un aumento en la incidencia de neumonía en pacientes con EPOC que reciben RELVARE® También se observó un aumento en la incidencia de neumonías que ocasionaron hospitalización. En algunos casos, estos eventos de neumonía fueron fatales (veáse Estudios clínicos y Reacciones Adversas). Los médicos deben mantenerse alerta ante la posibilidad del desarrollo de neumonía en pacientes con EPOC, ya que las características clínicas de dichas infecciones pueden imitar los síntomas de las exacerbaciones de EPOC. Los factores de riesgo de neumonía en pacientes con EPOC que reciben RELVARE® incluyen tabaquismo, pacientes con historia de neumonía previa, pacientes con índice de masa corporal < 25 kg/m2 y pacientes con un predicho VEF1 < 50%. Estos factores deben tomarse en consideración al prescribir furoato de fluticasona/vilanterol, y debe reevaluarse el tratamiento si se presenta neumonía. La incidencia de neumonía en pacientes con asma fue poco común. Los pacientes con asma que toman furoato de fluticasona/vilanterol 184/22 microgramos, pueden tener mayor riesgo de neumonía en comparación con aquellos que reciben furoato de fluticasona/vilanterol 92/22 o placebo. No se identificaron factores de riesgo. Efectos sobre la Capacidad para Manejar y Utilizar Maquinaria: No se han realizado estudios para investigar el efecto de RELVARE® sobre el desempeño al manejar o la capacidad de operar maquinaria. No se anticipa un efecto negativo sobre dichas actividades en base a la farmacología de furoato de fluticasona o de vilanterol.
Restricciones de uso durante el embarazo y la lactancia: Fertilidad: No existen datos de fertilidad en humanos. Los estudios en animales no mostraron un efecto de vilanterol ni de furoato de fluticasona sobre la fertilidad (véase Datos de Seguridad Preclínicos). Embarazo: Ha existido una exposición limitada en el embarazo en humanos. Los estudios en animales han demostrado toxicidad reproductiva después de la administración de agonistas beta2 y corticosteroides (véase Datos de Seguridad Preclínicos). La administración de furoato de fluticasona/vilanterol en mujeres embarazadas, solo debe considerarse si el beneficio esperado para la madre es mayor que cualquier posible riesgo para el feto. Lactancia: Existe información limitada sobre la excreción de furoato de fluticasona o vilanterol o sus metabolitos en la leche humana. Sin embargo, otros corticosteroides y agonistas beta2 pueden detectarse en la leche humana (véase Datos de Seguridad Preclínicos). No puede excluirse un riesgo para los recién nacidos que lactan/lactantes. Debe tomarse la decisión de no lactar o de suspender el tratamiento con RELVARE® tomando en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento para la madre.
Reacciones secundarias y adversas: Datos de estudios clínicos: Se utilizaron los datos de estudios clínicos grandes de asma y EPOC para determinar la frecuencia de las reacciones adversas asociadas con RELVARE®. En el programa de desarrollo clínico de asma, un total de 7,034 pacientes fueron incluidos en una evaluación integrada de reacciones adversas. En el programa de desarrollo clínico de EPOC, un total de 6,237 sujetos fueron incluidos en una evaluación integrada de reacciones adversas. Con excepción de neumonía y fracturas, el perfil de seguridad fue similar en pacientes con asma y EPOC. Durante los estudios clínicos, se observaron con mayor frecuencia neumonía y fracturas en pacientes con EPOC. Estos eventos adversos se enlistan por clase de sistema orgánico y frecuencia. Se ha utilizado la siguiente convención para la clasificación de las reacciones adversas: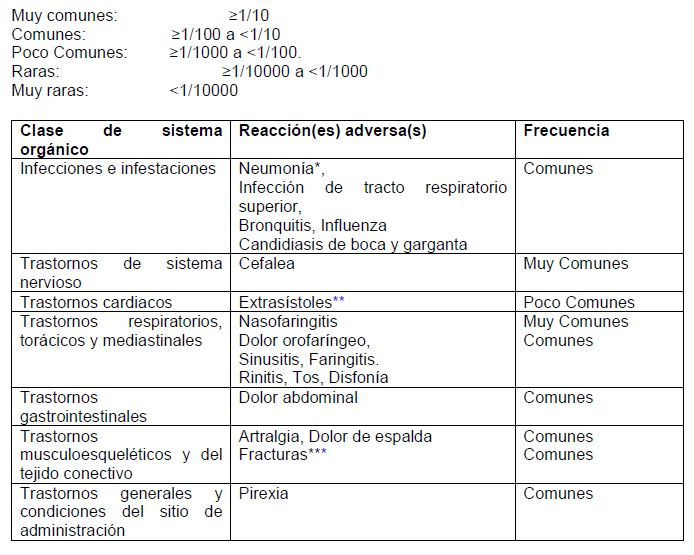
Descripción de las reacciones adversas seleccionadas: *Neumonía (véase Advertencias y Precauciones). En dos estudios duplicados de 12 meses realizados en un total de 3,255 pacientes con EPOC (al tamizado y post-broncodilatador, 45% del FEV1 predicho, desviación estándar (SD) 13% en promedio) quienes habían experimentado una exacerbación de EPOC en el año previo, se reportó una incidencia más alta de neumonía (6% - 7%) en pacientes que recibieron la combinación de furoato de fluticasona (concentraciones de 46, 92, y 184 microgramos)/vilanterol 22 microgramos en comparación con aquellos que recibieron vilanterol 22 microgramos sólo (3%). Ocurrió neumonía que requirió hospitalización en 3% de los pacientes que recibieron RELVARE® (todas las concentraciones) y en < 1% de los pacientes que recibieron vilanterol. En estos estudios, se reportaron nueve casos fatales de neumonía. De estos, siete se reportaron durante el tratamiento con RELVARE® 184/22 microgramos, uno durante el tratamiento con RELVARE® 92/22 microgramos, y uno después del tratamiento con vilanterol en monoterapia. En SUMMIT, un estudio multi-céntrico, randomizado (HZC113782), 16,568 sujetos recibieron furoato de fluticasona/vilanterol 100/25 microgramos, furoato de fluticasona 100 microgramos, vilanterol 25 microgramos, o placebo por un promedio de 1.7 años. Los sujetos tenían EPOC moderada (al tamizado y post-broncodilatador, 60% del FEV1 predicho, desviación estándar (SD) 6% en promedio) y una historia de, o un riesgo aumentado de enfermedad cardiovascular. Los eventos adversos de neumonía se anotan en la siguiente tabla.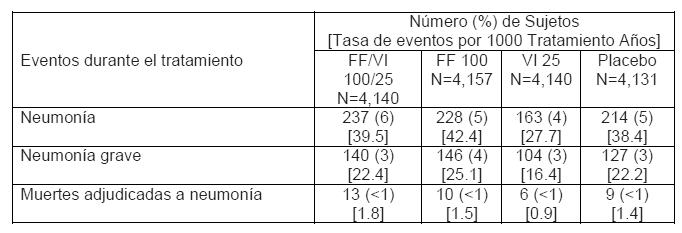
En un análisis integrado de 11 estudios en asma (7,034 pacientes), la incidencia de neumonía (ajustada para la exposición, debido a los números bajos y al número limitado de pacientes con placebo) observada con RELVARE® con una concentración de 92/22 microgramos (9.6/1000 años paciente) fue similar a la de placebo (8.0/1000 años paciente). Se observó una incidencia más alta de neumonía con la concentración de 184/22 microgramos (18.4/1000 años paciente) en comparación con la concentración de 92/22 microgramos. Pocos eventos de neumonía ocasionaron hospitalización con cualquier concentración, y no se observaron diferencias en la incidencia de eventos serios entre las dos concentraciones del tratamiento. ** Eventos Cardiovasculares (véase Advertencias y Precauciones). Para el estudio SUMMIT (véase arriba la descripción), los eventos adversos cardiovasculares se anotan en la siguiente tabla.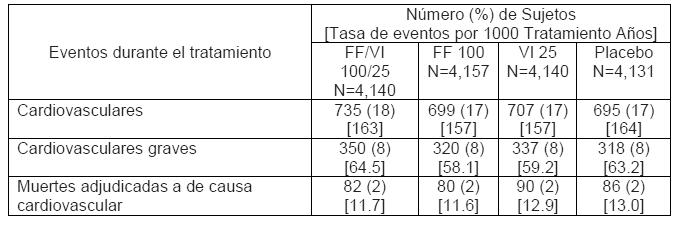
***Fracturas: En dos estudios duplicados de 12 meses con un total de 3,255 pacientes con EPOC, la incidencia de fracturas óseas en general fue baja en todos los grupos de tratamiento, con una incidencia más alta en todos los grupos de RELVARE® (2%) en comparación con el grupo de Vilanterol 22 microgramos ( < 1%). Aunque se observaron más fracturas en los grupos de RELVARE® en comparación con el grupo de vilanterol 22 microgramos, ocurrieron fracturas típicamente asociadas con el uso de corticosteroides (ej., compresión espinal/fracturas de vértebras toracolumbares, cadera y acetabulares) ocurrieron en < 1% de los grupos de tratamiento de RELVARE® y vilanterol. Para el estudio SUMMIT (véase arriba la descripción), las fracturas se anotan en la siguiente tabla.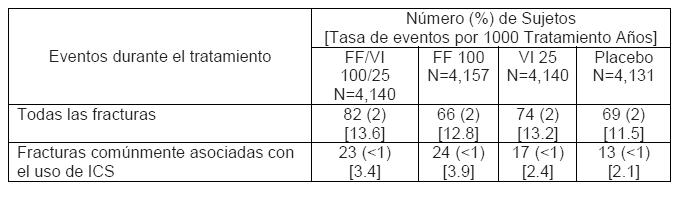
En un análisis integrado de 11 estudios en asma (7,034 pacientes), la incidencia de fracturas fue de < 1%, y usualmente se asociaron con trauma. Datos post comercialización: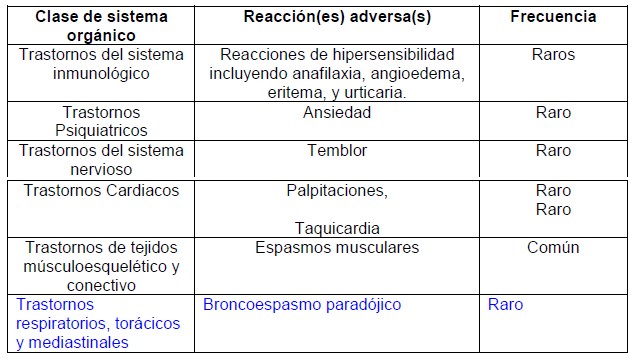
Interacciones medicamentosas y de otro género: Se consideran poco probables las interacciones farmacológicas mediadas por furoato de fluticasona o vilanterol a dosis clínicas, debido a las bajas concentraciones plasmáticas logradas después de la dosificación inhalada. Interacción con beta bloqueadores: Los bloqueadores beta adrenérgicos pueden debilitar o antagonizar los efectos de los agonistas beta2 adrenérgicos. Debe evitarse el uso concurrente de beta bloqueadores, tanto selectivos como no selectivos, a menos que existan razones de peso para su uso. Interacción con inhibidores del CYP3A4: Tanto el furoato de fluticasona como vilanterol son eliminados rápidamente mediante metabolismo de primer paso mediado por la enzima hepática CYP3A4. Se aconseja tener precaución al co administrar con inhibidores potentes del CYP 3A4 (ej. ketoconazol, ritonavir), ya que existe potencial de un aumento en la exposición sistémica, tanto a furoato de fluticasona como a vilanterol, lo que podría ocasionar reacciones adversas (véase Farmacocinética). Interacción con inhibidores de la glicoproteína P: Tanto el furoato de fluticasona como el vilanterol son sustratos de la glicoproteína P (P-gp). Un estudio de farmacología clínica realizado en sujetos sanos a quienes se coadministró vilanterol con el inhibidor potente de la P-gp y moderado del CYP3A4, verapamilo, no mostró ningún efecto significativo sobre la farmacocinética de vilanterol. No se han realizado estudios de farmacología clínica con un inhibidor específico de la P-gp y furoato de fluticasona.
Alteraciones en los resultados de pruebas de laboratorio: No reportadas.
Precauciones generales en relacion con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad: Datos de seguridad pre clínicos: Los efectos farmacológicos y toxicológicos observados con furoato de fluticasona o vilanterol en estudios no clínicos, fueron aquellos típicamente asociados con los glucocorticoides o los agonistas beta2. La administración de furoato de fluticasona combinado con vilanterol, no resultó en ninguna toxicidad nueva significativa. Carcinogénesis/mutagénesis: El furoato de fluticasona no fue genotóxico en un grupo estándar de estudios, y no fue carcinogénico en estudios de inhalación durante toda la vida en ratas o ratones con exposiciones similares a las observadas con la dosis máxima recomendada en humanos, con base en el AUC. Los estudios de toxicidad genética indicaron que Vilanterol no representa un riesgo genotóxico para los humanos. De manera consistente con los hallazgos con otros agonistas beta2 en los estudios de inhalación durante toda la vida, vilanterol ocasionó efectos proliferativos en tracto reproductivo de ratas y ratones hembra, y en glándula pituitaria de ratas. No se observó un aumento en la incidencia de tumores en ratas ni en ratones con exposiciones de 2 ó 30 veces, respectivamente, mayores a las observadas con la dosis humana recomendada, en base al AUC. Toxicología Reproductiva: Los efectos observados después de la administración mediante inhalación de Furoato de Fluticasona combinado con Vilanterol en ratas, fueron similares a los observados con Furoato de Fluticasona solo. Furoato de Fluticasona no fue teratogénico en ratas ni en conejos, pero retrasó el desarrollo en ratas, y ocasionó abortos en conejos con dosis maternas tóxicas. No se observaron efectos sobre el desarrollo en ratas con exposiciones aproximadamente del triple a las observadas con la dosis máxima recomendada en humanos, en base al AUC. Vilanterol no fue teratogénico en ratas. En estudios de inhalación en conejos, Vilanterol ocasionó efectos similares a los observados con otros agonistas beta2 (paladar hendido, párpados abiertos, fusión de esternebras, y flexión/malrotación de extremidades). Cuando se administró por vía subcutánea, no se observaron efectos con exposiciones 84 veces mayores a las observadas con la dosis máxima recomendada en humanos, en base al AUC. Ni Furoato de Fluticasona ni Vilanterol ocasionaron efectos adversos sobre la fertilidad ni sobre el desarrollo pre y post natal en ratas.
Dosis y via de administración: RELVARE® solo es para uso mediante inhalación. RELVARE® debe administrarse una vez al día en las mañanas o en la tarde, pero a la misma hora cada día. Después de la inhalación, el paciente debe enjuagar su boca con agua, sin tragarla. Asma: Debe advertirse a los pacientes que RELVARE® debe utilizarse de forma regular, incluso aunque no presenten síntomas. Si surgen síntomas en el periodo entre dosis, debe tomarse un agonista beta2 inhalado, de acción corta, para obtener alivio inmediato. Los pacientes deben ser reevaluados de forma regular por un profesional de la salud, para que la concentración de furoato de fluticasona/vilanterol que están recibiendo se mantenga óptima, y sólo cambie por recomendación médica. Poblaciones: Adultos y adolescentes de 12 años de edad y mayores: La dosis recomendada de RELVARE® es: Una inhalación RELVARE® de 92/22 microgramos una vez al día o Una inhalación de RELVARE® de 184/22 microgramos una vez al día. Debe considerarse una dosis inicial de RELVARE® de 92/22 microgramos en pacientes que requieren una dosis de baja a mediana de un corticosteroide inhalado en combinación con un agonista beta2 de larga duración. RELVARE® 184/22 microgramos debe considerarse en pacientes que requieren una dosis más alta de corticosteroides inhalados en combinación con un agonista beta2 de larga duración. Si los pacientes no se controlan de forma adecuada con RELVARE® 92/22 microgramos, considere aumentar la dosis a 184/22 microgramos, lo cual puede proporcionar una mejoría adicional para el control del asma. Niños: No se ha establecido la seguridad y eficacia de RELVARE® en niños menores de 12 años de edad. EPOC Poblaciones Adultos: La dosis recomendada de RELVARE® es: Una inhalación de RELVARE® de 92/22 microgramos una vez al día. RELVARE® de 184/22 microgramos no está indicado en pacientes con EPOC. Niños: El uso en niños no es relevante debido a la indicación de EPOC de este producto. Población especial: Asma y EPOC Ancianos: No se requiere ajustar la dosis en pacientes mayores de 65 años (ver Farmacocinética - Poblaciones Especiales de Pacientes). Insuficiencia renal: No se requiere ajustar la dosis en pacientes con insuficiencia renal (ver Farmacocinética). Insuficiencia hepática: Un estudio de farmacología clínica en sujetos con insuficiencia hepática leve, moderada y severa, mostró un aumento del triple en la exposición sistémica a furoato de fluticasona (tanto Cmax como AUC) (véase Farmacocinética). Debe tenerse precaución al administrar en pacientes con insuficiencia hepática, ya que los pacientes con insuficiencia hepática pueden tener mayor riesgo de reacciones adversas sistémicas asociadas co